
CUESTIONES PREVIAS
¿Qué es la cromatografía?
La cromatografía es un método químico de separación para la caracterización de mezclas complejas. La cromatografía tiene aplicaciones en todas las ramas de la ciencia, como por ejemplo, en el principio de retención selectiva, cuyo objetivo es separar los distintos componentes de una mezcla, permitiendo identificar y determinar las cantidades de dichos componentes.
¿Por qué obtenemos un cromatograma?
Cuando realizamos una secuenciación de ADN, obtenemos información sobre el orden de los nucleótidos de un fragmento de ADN, los cuales provienen, por ejemplo, de una PCR. En la investigación biomédica, la secuenciación se realiza para conocer las variaciones en la secuencia de un fragmento de ADN o un gen específico de un individuo. Esto es esencial para realizar un diagnóstico de alguna enfermedad.
Existen diferentes métodos de secuenciación, donde también destaca la Secuenciación Sanger (ver pubicación: ¿cómo clonar el gen de la insulina humana?). Los servicios de secuenciación de ADN envían los resultados de sus análisis como secuencias en formato FASTA y, además, incluyen, o deberían incluir siempre, el cromatograma que produce el equipo junto con la información de calidad de la secuencia base a base.
¿Por qué obtenemos un cromatograma de mala calidad?
Podemos encontrar diferentes tipos de cromatograma de mala calidad. Por eso, siempre es conveniente analizar los picos obtenidos y las bases a las que se corresponden. Además, se recomienda secuenciar dos veces como mínimo, para poder comparar las secuencias obtenidas, con el objetivo de minimizar los errores que se presentan a continuación:
– Un cromatograma que presenta una señal inicial y a continuación decae de forma significativa puede deberse a una insuficiente sensibilidad por la muestra de ADN. Es decir, que haya poca muestra de ADN, pocos cebadores, cebadores poco específicos, presencia de contaminantes, etc. Se observaría como el cromatograma representado en la figura 1
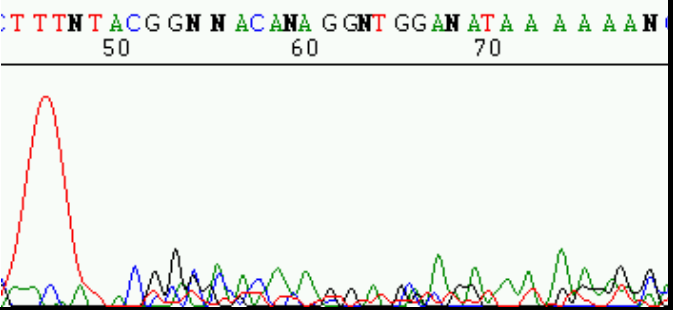
– Un cromatograma donde se observan picos definidos y separados, pero con ruido de fondo. Esto podría deberse a que el cebador tenga afinidad por un sitio de unión secundario, aunque en menor medida que por el sitio principal. También puede surgir un cromatograma como el de la figura 2 por errores en el proceso de purificación del ADN.

Para más información sobre la evaluación de datos de cromatograma, así como posibles soluciones a los errores encontrados, pulsar aquí.
PASO 1. ANÁLISIS DE LA SECUENCIA. Visualización y evaluación de la secuencia
VISUALIZACIÓN
Para analizar un cromatograma podemos recurrir a diferentes programas, que nos permiten una visualización y corrección de la secuencia. Algunos ejemplos útiles se muestran a continuación:
– Chromas. Este será el recurso que se utilizará para este procedimiento.
– Sequence Scanner Software v1.0
– FinchTV (Libre para Linux, PCs, y MACs)
EVALUACIÓN
¿HAY ERRORES OBVIOS EN LA SECUENCIA DE NUCLEÓTIDOS?
Al analizar la secuencia, nos encontramos muchas bases sin resolver, que en el programa se presentarán como una «N», debido a diferentes situaciones:
– Inserción errónea de nucleótidos en la secuencia
– La superposición de picos y picos mal espaciados
– Ruido de fondo
– Pérdida de resolución en la secuenciación. A medida que avanza la secuenciación de las bases, los picos se ensanchan y pierden calidad de resolución
– Mala calidad de los extremos en el proceso de secuenciación. Cuando se secuencia un fragmento de ADN, las primeras 25-30 pb no se consiguen resolver, por eso es muy importante el diseño de cebadores para amplificar el ADN en la reacción de PCR. La eliminación de los extremos sólo se refiere al extremo izquierdo de la cadena. El extremo derecho será eliminado según el criterio del programa.
ANÁLISIS SUPERFICIAL DE LA SECUENCIA
Tal y como se ha descrito, lo primero que debemos hacer es una evaluación de la secuencia, para determinar ante qué situación nos encontramos y qué errores tenemos por delante. En las siguientes figuras se muestran ejemplos del análisis superficial realizado de nuestra secuencia problema.



PROCEDIMIENTO
ELIMINACIÓN DE EXTREMOS SIN RESOLVER
- Selecciona la base N anterior a la primera base diferente de N
- En la barra de herramientas: Edit –> Set left Trim
- Quedarán seleccionadas todas las bases N del extremo izquierdo de la secuencia (figura 6).
- En la barra de herramientas: Edit –> Clear left Trim
- Quedarán eliminadas las bases seleccionadas.

CORRECCIÓN DE BASES SIN RESOLVER EN LA SECUENCIA O BASES MAL RESUELTAS
En algunas situaciones, el programa no ha conseguido resolver alguna base y coloca una «N». En otras ocasiones, atribuye bases erróneas a los picos o deja espacios sin resolver donde existe un pico acusado.
Esto se ve de manera significativa en las primeras 160 bases, donde la abundancia de N, la presencia de picos sin resolver (espacios entre bases), picos que se han resuelto erróneamente (bases que no se corresponden a esos picos), etc. nos obliga a hacer un set left trim de estas primeras 160 bases y así deshecharlas.

PASO 2. COMPARACIÓN OBJETIVA. Alineamiento local
INTRODUCCIÓN
En la corrección del cromatograma es importante no cambiar bases de una forma subjetiva y sin la seguridad de que estemos colocando bases en lugares donde se encontrarían, atribuyendo picos a bases correctas, etc. Un ejemplo de esta situación se observa en la figura 8.

Por eso, es necesario comparar la secuencia que estamos corrigiendo con otras ya secuenciadas, para encontrar identidades o discrepancias. Esto puede realizarse mediante:
- Visualización en NCBI
- Re-secuenciación
En este caso realizaremos un alineamiento local en NCBI.
PROCEDIMIENTO
ANÁLISIS DEL ALINEAMIENTO LOCAL
Al exportar la secuencia problema (Query) obtenemos un diagrama de distribución del alineamiento local (figura 9), al que se le han atribuido puntuaciones, dependiendo de las bases alineadas. Se recomienda tratar esta infromación con cautela y preguntarnos ¿por qué no aparece puntuación en algunas zonas de la secuencia?
La respuesta a la pregunta es la siguiente: Podemos encontrarnos diversas situaciones que provoquen que el alineamiento local del NCBI no lo interprete por válido y, por lo tanto, no lo muestre. Puede darse el caso de que haya fallos en la secuencia, en cuyo caso habría que corregir, que invaliden este alineamiento o puede suceder que sean dos secuencias completamente distintas. De cualquier modo, con una simple visión de este diagrama no podríamos saber a qué situación nos enfrentamos. Por eso, el alineamiento local nos sirve como referencia pero interpretándolo con cuidado.

Observando el diagrama, vemos que todos los alineamientos encontrados presentan una alta puntuación. Sin embargo, la primera secuencia encontrada es la que presenta mayor alineamiento local. En una vista más detallada de la secuencia encontrada (subject) vemos que corresponde al factor de transcripción BHLH 75 de la fresa, y que presenta un 97% de identidad con la secuencia Query (figura 10)

En principio, parece que el alineamiento local ha abarcado gran cantidad de secuencia, aunque no muestra el alineamiento con las últimas 79 bases (868 -789). Para comparar esta última parte de la secuencia cambiaremos la vista de la región de la secuencia subject en NCBI, amlpiando +100 bases (885 + 100 bases) (figura 11)

CORRECCIÓN DE BASES NO ALINEADAS
A continuación, observamos las bases que no han coincidido con el alineamiento local y tratamos de corregirlas en el cromatograma. De esta manera, en aquellos picos donde se presentaban dudas por la posible existencia de una base, o por la superposición de picos, podemos exclarecer qué elemento es el correcto.
Es importante tener en cuenta que hay ocasiones en las que la secuencia subject nos indica que hay una base nitrogenada pero en el cromatograma se observa claramente la existencia de una diferente. Esto puede verse en un ejemplo en la figura 12

Al finalizar las correcciones posibles obtenemos una nueva secuencia a la que llamaremos secuencia Query_1
PASO 3. COMPARACIÓN DETALLADA. ALINEAMIENTO GLOBAL
INTRODUCCIÓN
Como se mencionó anteriormente, para una comparación detallada de las secuencias ha sido necesario ampliar la ventana de bases en la secuencia subject, debido a que no se estaban teniendo en cuenta las últimas 79 bases de la secuencia Query_1.
En esta sección es necesario exportar ambas secuencias (Query_1 y subject) en formato FASTA, para su comparación global a través de bases de datos que lo permitan. Algunos ejemplos de bases de datos que realizan alineamiento global son:
– EMBL-EBI
– Uniprot
– NCBI
En este caso realizaremos un alineamiento global en EMBL-EBI
PROCEDIMIENTO
SECUENCIAS EN FORMATO FASTA
Para realizar el alineamiento global de ambas secuencias es necesario exportarlas en formato FASTA. Es importante no dejar muchas bases sin resolver (N) en la secuencia prblema (Query_1) porque de lo contrario, no se analizará bien. Además, para resolver este alineamiento de una forma más exacta, la secuencia subject en formato FASTA debe incluir las últimas ~100 bases, para compararlas con las últimas 79 bases de nuestra secuencia.
ANÁLISIS DEL ALINEAMIENTO GLOBAL
Obtenemos ambas secuencias alineadas desde la base número 162 (se corresponde con base nº1 en EMBL-EBI) hasta la base 868 (se corresponde con base nº 707 en EMBL-EBI) de la secuencia de Query_1. Y desde la base 256 (se corresponde con base nº 1 ) hasta la base 985 (se corresponde con la base 730) de la secuencia de Subject (figura 14).

En este alineamiento global se ha obtenido un 87,2% de identidad. Este resultado es inferior al obtenido en el alineamiento local, pero resulta lógico al tratarse de un alineamiento que está teniendo en cuenta toda la longitud de la secuencia Query_1. Por lo tanto, a pesar de ser un resultado inferior, es alto al referirse a toda la secuencia.
Si observamos las bases señaladas en la figura, podemos ver el alineamiento en la zona de la secuencia que no se estaba teniendo en cuenta en el alineamiento local. En este momento, es posible que se puedan corregir algunas bases que no estén alineadas.
Al corregir algunas bases que no están alineadas en el alineamiento global, obtenemos una nueva secuencia a la que llamaremos Query_2
NUEVA SECUENCIACIÓN GLOBAL
Debido a que se han realizado nuevas correcciones en algunas bases del final de la secuencia Query_1 tras el alineamiento global, se volverá a hacer un alineamiento con la misma secuencia Subject. Además, así podremos comparar el porcentaje de identidad al realizar correcciones en la región final de la secuencia, con el obtenido en el apartado anterior, que solo tenía en cuenta las pocas correcciones que se pudieron hacer en el alineamiento local.
De esta manera, obtenemos la figura 15, donde muestra un porcentaje de identidad igual a 87,7%. Podemos comprobar que, a pesar de no ser una gran diferencia, pues solo hemos corregido unas pocas bases, en la secuencia Query_2 obtenemos mayor porcentaje de identidad.

PASO 4. SECUENCIA PEPTÍDICA. INFORMACIÓN ADICIONAL DE LA SECUENCIA
INTRODUCCIÓN
Con el objetivo de obtener información adicional sobre la secuencia nucleotídica corregida en el paso 3, se obtendrá la secuencia protéica, si es que la hubiera, y se buscarán posibles identidades o discrepancias.
En esta sección se pueden utilizar varias bases de datos (por ejemplo, Uniprot). En este caso se recurrirá a BLAST X de NCBI para encontrar la secuencia protéica y posibles alineamientos.
PROCEDIMIENTO
ANÁLISIS DEL ALINEAMIENTO DE SECUENCIAS PEPTÍDICAS
Al igual que para el alineamiento local, al introducir la secuencia peptídica Query_2, obtenemos un diagrama de distribución del alineamiento protéico (no se muestra en figura), el cual atribuye diferentes puntuaciones a las zonas alineadas de la secuencia.
Alineamiento de secuencia peptídica Query_1
Al acceder a la secuencia con mayor puntuación de alineamiento predicha (Subject) observamos que se trata de la isoforma X1 del factor de transcripción BHLH 75 (Figura 16), con un 89% de identidad.

Alineamiento de secuencia peptídica Query_2
Al acceder a la secuencia con mayor puntuación de alineamiento predicha (Subject) observamos que se trata de la isoforma X1 del factor de transcripción BHLH 75 (Figura 17), con un 89% de identidad. Resulta curioso pero ambas presentan el mismo porcentaje de identidad.

Debido a que ambas secuencias (Query_1 y Query_2) presentan el mismo porcentaje de identidad con la secuencia del factor de transcripción BHLH 75 isoforma 1x, se realizará a continuación un alineamiento entre las dos secuencias Query, a través de un alineamiento global de secuencia con NCBI. En este nuevo alineamiento, la secuencia «Query» será la secuencia Query_1, mientras que la secuencia «subject» será la secuenci Query_2.
Alineamiento de secuencias peptídicas sin corregir y corregidas entre sí
El resultado de este alineamiento, tal y como se esperaba, presenta un porcentaje de identidad del 100%, debido a que son exactamente la misma secuencia (figura 18). Es decir, a pesar de haber corregido algunas bases entre Query_1 y Query_2 (incluso, en algunos casos, haber eliminado bases), hemos obtenido exactamente la misma secuencia protéica. Esto resulta como consecuencia de que el código genético es degenerado y, por lo tanto, algunas «mutaciones» en la secuencia (no estamos realizando mutaciones, simplemente correcciones), pueden dar lugar a la misma secuencia protéica.

FIN. Curiosidades y valoración personal
Se puede concluir que la secuencia del cromatograma corresponde al factor de transcripción BHLH 75 de la fresa. Además, al introducir la secuencia nucleotídica en la base de datos ENSEMBL, podemos observar que, realizando un BLAT, se encuentra un 100% de identidad con una región de 22 nucleótidos del cromosoma 6 de Homo sapiens. Es decir, en el cromosoma 6 de las personas se encuentra un fragmento de ADN que presenta la misma secuencia de nucleótidos que el factor de transcripción de una fresa. (figura 19)

La región del cromosoma 6 donde se encuentra el fragmento de ADN de la secuencia del cromatograma (en concreto de la secuencia Query_2) se muestra en la figura 20. Esta región presenta 7 transcritos, de los cuales 4 son intrones y 3 son proteínas.

Desde mi punto de vista, me gustaría destacar la importancia del correcto manejo de las bases de datos y las herramientas bioinformáticas como parte de un buen proyecto bioquímico, biológico, etc. Dichas herramientas pueden facilitar el trabajo de investigación, pero también pueden hacer que fracasen si no se saben utilizar. Por ejemplo, la interpretación errónea de un cromatograma cuando se obtiene una secuencia nucleotídica puede resultar en un error de experimento.
Por este motivo, es necesario aprender a manejar y a obtener la información, así como a realizar una valoración de la misma y poder corregirla en caso de fallos. Y esto solo se consigue haciéndolo.
This Post Has 0 Comments