
Cuestiones previas
Definición
Se han reconocido diferentes moléculas y sistemas que se encargan de silenciar la expresión y se encuentran especialmente en el PTGS. Dentro de estas moléculas se encuentran proteínas “Dicers”, argonautas, con actividad RNAsa, miRNA (micro ARN de interferencia), siRNA (ARN pequeño de interferencia) con variaciones como nat-siRNA (inducido durante situaciones de estrés) y ra-siRNA para la transcripción de secuencias repetidas. Adicionalmente, se encuentran complejos que se acoplan con estos elementos, como los RISC (Complejo de Silenciamiento inducido por ARN) actúando a nivel citoplasmático acoplando el siRNA producido, y el sistema RIST (Complejo de Silenciamiento Transcripcional Inducido por ARN) cuya actividad se localiza en el núcleo celular.
El silenciamiento de ARN es un mecanismo regulador de genes que actúa inactivando la transcripción (silenciamiento génico transcripcional, TGS) o mediante la activación de un proceso de degradación del ARN específico de la secuencia (silenciamiento génico postranscripcional, PTGS / interferencia de ARN, ARNi). Existe una relación en el mecanismo de TGS y PTGS, pero en esta sección se recurrirá a los fenómenos relacionados con PTGS/ARNi
Aunque tanto los ARN de pequeño tamaño (sRNA) como los RNA que codifican proteínas (mRNA) poseen variaciones, los sRNA pueden regular eficazmente la expresión génica, el empalme de genes, las modificaciones de nucleótidos y el transporte de proteínas. Las diferencias y similitudes entre sRNAs y mRNAs se presentan en la tabla 1.
| Propiedad | ARN no codificante | ARN codificante |
| Longitud | 20-30 nt (ARN pequeños procesados) 62-303 nt (precursores de plantas) |
polinucleótidos |
| Ubicación de la síntesis | Núcleo y citoplasma | Núcleo y citoplasma |
| ARN polimerasa | ARN polimerasa II y IV | ARN polimerasa II |
| Síntesis de proteínas | No | Sí |
| Unión a proteína Argonauta | Sí | No |
| Tasa de degradación | Menos estable | Más estable |
| Marcos de lectura abiertos | Ausente | Presente |
| Respuesta a mutaciones puntuales | Menos sensible | Más sensible |
| Efecto de mutaciones puntuales | Más drástico | Menos drástico |
| Funciones | Silenciamiento génico transcripcional y postranscripcional | Expresión de genes |
| Tipos identificados | miARN, siARN, tasiARN, rasiARN, vsiARN, piARN | ARNm |
Características del silenciamiento del ARN en plantas
Para desarrollar el silenciamiento de un gen es importante entender las características básicas de este proceso, así como una visión general de los componentes fundamentales del ARNi.
En las plantas, la historia del silenciamiento del ARN se desarrolló por casualidad durante una búsqueda de flores de petunia transgénicas que se esperaba que fueran más moradas. Jorgensen acuñó el término cosupresión para describir la pérdida de ARNm, tanto del endo como del transgén en sus experimentos.
Los componentes comunes del proceso de silenciamiento de ARN, independientemente de si se trata de un mecanismo de PTGS, ARNi, etc., son que 1) el inductor es el dsRNA, 2) el ARN diana se degrada en una forma dependiente de homología y, 3) La maquinaria degradativa requiere un conjunto de proteínas similares en estructura y función en la mayoría de organismos
El producto de degradación de ARN en el proceso de PTGS es un pequeño ARN (siARN) de ~25 nucleótidos con polaridad sentido y antisentido, los cuales se forman y se acumulan como bicatenarios. De esta manera, los siARN inducen la degradación (si la complementariedad es absoluta) del ARNm diana.
PROPAGACIÓN SISTÉMICA DEL SILENCIAMIENTO GÉNICO
La persistencia y duración de la señal de silenciamiento en las plantas parece estar relacionado con los dominios helicasa y/o RNasa en algunas especies. Se ha comprobado que en humanos existen unas proteínas SMG que desenrollan el dsRNA y facilitan la amplificación de la señal de silenciamiento, así como la persistencia de su efecto. Sin emabrgo, estas proteínas parecen no estar presentes en plantas ni hongos.
Además, es importante tener en cuenta la transmisión sistémica de la señal de silenciamiento por la planta. En algunos trabajos se ha visto que la señal de silenciamiento se transmite de forma no metabólica, viajando entre las células, a través de los plasmodermos, incluso recorriendo largas distancias a través del floema, mediante una señal difusible de un gen.
Se identificó un locus sistémico deficiente en interferencia de ARN (SID) que se requiere para transmitir los efectos del gen en las células. Basasándose en la estructura de SID1 (mutante SID) se sugirió que podría actuar como canal para la importación o exportación de una señal de iARN sistémica, o que podría estar relacionado con la endocitosis de la señal sistémica de iARN, quizás funcionando como receptor. Así mismo, la presencia de homólogos SID en humanos y ratones podría explicar algunas características sistémicas de iARN en mamíferos.
COMPONENTES DEL COMPLEJO DE SILENCIAMIENTO
Algunos de los componentes identificados sirven como iniciadores, mientras que otros sirven como efectores, amplificadores y transmisores del proceso de silenciamiento génico.
Se ha identificado una enzima similar a la RNasa III capaz de producir fragmentos de 22 nucleótidos, involucrado en la iniciación de siRNA. Debido a su capacidad para digerir fragmentos de doble cadena de ARN (dsRNA), se le denominó DICER. Estas nucleasas se conservan evolutivamente en gusanos, moscas, hongos, plantas y mamíferos, por lo que es probable que sea funcionalmente similar en distintas especies.
Los dsARN serán reconocidos y procesados por DICER, para dar lugar a los siRNA, con extremos 5′ fosforilados y dos nucleótidos en el extremo 3′ volatilizados. . Después del procesamiento, los siRNA de plantas a menudo se modifican por 2′-O-metilación a las 3′termina por HUA ENHANCER 1 (HEN1) para conferir estabilidad y evitar la degradación por nucleasas.
Además, los ARN pequeños se asocian con nucleasas específicas de secuencia, y sirven como guías para mensajes específicos de destino, basados en el reconocimiento de la secuencia. Se ha observado una actividad nucleasa específica de secuencia en extractos celulares responsables de la degradación de ARNm diana, a la cual se le denominó actividad del complejo de silenciamiento inducido por ARN (RISC).
Uno de los componentes protéicos de este complejo pertenece a la familia de proteínas Argonauta, denominada argonauta 2 (AGO2). Los miembros de la familia argonauta participan en procesos de silenciamiento de genes, así como en la regulación del desarrollo de diferentes especies.
Por otro lado, parece ser común entre organismos eucariotas el proceso de eliminación de ARN aberrante en las células. Sin embargo, son diferentes las proteínas específicas que llevan a cabo este proceso entre organismos. La ARN helicasa MUT6 puede estar involucrada en la degradación de ARN aberrantes mal procesados y, por lo tanto, podría ser parte de un sistema de vigilancia relacionado con RNAi.
MECANISMO PARA EL SILENCIAMIENTO GÉNICO
Como ya se ha introducido anteriormente, el proceso de silenciamiento génico mediado por iARN consiste en el reconocimiento y procesamiento de dsRNA largo, con la ayuda de Dicer. La asimetría del dúplex de siRNA determina qué hebra ingresa a Dicer. El siRNA originado a partir de Dicer es un intermediario, que será reconocido por el complejo RISC para seleccionar el ARNm a degradar según complementariedad (figura 1).
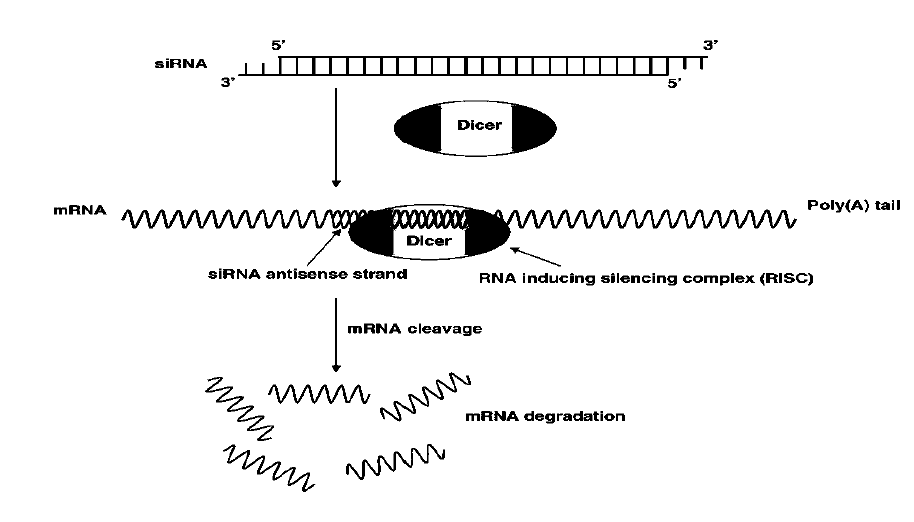
Los siRNA están estrechamente relacionados con los miRNA pero difieren en cuanto a su origen, estructura, proteína efectora asociada y modo de acción. Las diferencias entre miRNAs y siRNAs se presentan en la tabla 2.
| Propiedad | miRNA | siRNA |
| Definición | Reguladores de genes endógenos | Defensores de la integridad del genoma en respuesta a ácidos nucleicos extraños o invasivos |
| Longitud | 20-22 nt | 21-24 nt |
| Precursores | ARNss en forma de horquilla | ARNbc largos |
| Naturaleza de los precursores | Gen precursor endógeno del genoma del huésped | Transposones, transgenes, elementos repetidos o virus, es decir, precursores exógenos |
| Modo de acción | Degradación de ARNm, represión traduccional. | Metilación del ADN, modificación de histonas y degradación del ARNm |
| Argonauta requerido | AGO1, AGO10 | AGO1, AGO4, AGO6, AGO7 |
| Mecanismo de regulación génica | Solo postranscripcional | Transcripcional y postranscripcional. |
| Complementariedad con secuencias diana | Parcial o totalmente complementario | Completamente complementario |
| Funciones | Desarrollo celular y diferenciación celular, regulación de procesos de desarrollo, respuestas al estrés biótico y abiótico | Defensa contra transposones y virus, adaptación al estrés. |
TECNOLOGÍAS ÚTILES PARA EL SILENCIAMIENTO DE UN GEN
CUÁLES, CÓMO Y POR QUÉ
Ya se ha introducido la tecnología que se desarrollará en esta entrada del blog (ARN de interfrencia o RNAi). Sin embargo, se pueden mencionar algunas otras que también son útiles, así como su funcionamiento y mecanismo de silenciamiento, y sus ventajas e inconvenientes en el silenciamiento del gen de la polifenol oxidasa.
ADN antisentido
La tecnología del ADN antisentido se basa en la especificidad de una cadena de ADN con su complementaria. La replicación no sería posible sin dicha complementariedad, debido a que cada hebra sirve como plantilla para reconstruir a su pareja, y el resultado son dos dúplex idéntico.
Además, para la síntesis de proteínas es necesaria dicha complementariedad de bases, ya que el intermediario entre los genes y su expresión en la célula es el ARNm, formado a partir de la complementariedad con la cadena antisentido de ADN (sustituyendo la base timina (T) por uracilo (U)). La hebra antisentido de ADN en los genes es la plantilla parael ARNm.
Pero, ¿qué pasa con la hebra de ADN sentido? ¿Produce ARN antisentido y, de ser así, qué
hace este ARN?
En 1981 Tomizawa estudió la replicación de plásmidos en una bacteria llamada ColE1 y descubrió que la disponibilidad de cebadores de ARN no estaba controlada por su concentración total sino más bien por la proporción de cebadores y moléculas inhibidoras específicas. Estos inhibidores eran los productos antisentido de las cadenas de ADN con sentido. Debido a la complementariedad entre el ADN con sentido y el ADN antisentido, sus productos de reacción también eran complementarios, por lo que así funcionaba la inhibición.
El ARN con sentido y el ARN antisentido pueden hibridarse entre sí. En este estado duplicado, el cebador de ARN no puede iniciar la replicación del ADN porque no puede emparejarse con el origen del plásmido (figura 2). El silenciamiento génico inducido por ARN antisentido también se explicó proponiendo que la síntesis de ARN estaba cebada en el ARNm por ARN antisentido, lo que da lugar a un dsRNA, actuando como sustratos para la degradación dependiente de Dicer.
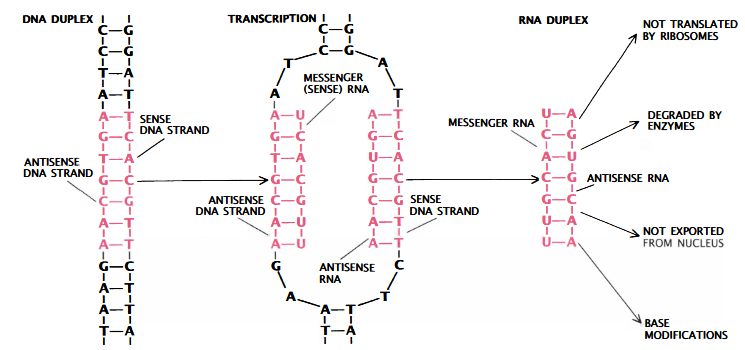
De esta manera, se podría dirigir el ARN antisentido contra cualquier gen clonado y tal vez
inhibir la traducción de su ARNm. Así, se podrían inactivar genes específicos, como le de la polifenol oxidasa.
Utilizando la tecnología del ADN recombinante (ver enlace para más información sobre ADN recombinante) se pueden crear elementos genéticos artificiales, conocidos como vectores de expresión, para producir ARN antisentido en una célula (figura 3).

Según la figura 3:
- Se aísla una molécula de ARNm.
- Este ARNm sirve como plantilla para crear una cadena de ADNc (es ADN antisentido)
- El ADN complementario se separa
- El ADN complementario sirve como plantilla para crear un ADN con sentido y formar un dúplex de ADN.
- y 6. Luego, un plásmido se puede cortar con enzimas de restricción cerca de la secuencia promotora

Según la figura 4:
7. El dúplex de ADN se inserta en el plásmido, formando así el vector de expresión. Cuando el promotor inicie la transcripción, se crearán copias del ARN mensajero original (azul)
8. Luego, se vuelve a cortar el dúplex de ADN que fue añadido con enzimas de restricción
9. Posteriormente, se vuelve a insertar dicho dúplex de ADN en el plásmido, pero en dirección opuesta. Durante la transcripción, se expresará el ARN antisentido (amarillo).
CRISPR/Cas9
En los últimos años han surgido distintos métodos de modificación dirigidos que utilizan meganucleasas, nucleasas con dedos de zinc (ZFN), nucleasas efectoras similares a activadores de transcripción (TALEN) y repeticiones de palíndromo cortas agrupadas regularmente interespaciadas (CRISPR)/proteína 9 asociada a CRISPR (Cas9).
La tecnología de CRISPR/Cas9 se explica en el vídeo 1. Para más información sobre el descubrimiento y aplicaciones de esta tecnología, visita este enlace.
TALEN
Las nucleasas efectoras similares a activadores de la transcripción (TALEN) constan de un dominio de unión y de nucleasa que generalmente se usan en forma de un par. El dominio de unión se une específicamente a una secuencia de ADN, mientras que el dominio de nucleasa crea roturas de doble cadena (DSB) que se utilizan posteriormente para la unión de extremos no homólogos o la reparación por recombinación homóloga. La creación de DSB es el principio de esta tecnología que se puede utilizar para la adición, eliminación y modificación de genes en el ADN diana.
Además de la actividad de la nucleasa, las proteínas TALE (efectores similares a activadores de la transcripción) también se han utilizado junto con otros dominios efectores para diferentes propósitos, como activación de genes, represión de genes, modificaciones epigenéticas, etc.
Las proteínas TALE están compuestas por monómeros TALE, por una región N-terminal, región C-terminal, región NLS (señal de localización nuclear), región AD (dominio de activación) y una región TS (señal T3S). Los monómeros TALE son repeticiones en tándem de unos ~34 aminoácidos. Cada repetición de aminoácido se une específicamente a una base del ADN. En concreto, la unión a la base de ADN se realiza a través del aminoácido en la posición 12 y 13. A estos aminoácidos en la posición 12 y 13 se les conoce como di-residuos variables repetidos (RVDs) (figura 5).

Además, la proteína TALE presenta un código de unión al ADN. Existen 4 RVDs que se unirán de manera específica a una base de ADN. Estas son NN, NG, NI, HD y se unen a G, T, A, C respectivamente (figura 6).

CRISPR/Cas9 vs TALENs vs RNAi
El siguiente vídeo (vídeo 2) resume el mecanismo, el diseño experimental, la eficiencia, los ejectos fuera del objetivo, la flexibilidad y aplicaciones de estas tres técnicas, que ya se han introducido anteriormente. De esta manera, se puede obtener una comparación entre estas tecnologías útiles para el silenciamiento y manipulación del genoma.
DISEÑO DE ARNi
INTRODUCCIÓN
En esta sección se plantearán otros aspectos a tener en cuenta en el diseño del ARNi de interés
1. Secuencias de 21 nucleótidos en el ARNm objetivo que comiencen con un dinucleótido AA.
Seleccione regiones de ADN para servir como plantillas para dsRNAs de 400 pb o más, correspondiente a un gen de interés. Debe evitar los intrones y, para minimizar los potentes efectos fuera del objetivo, la secuencia objetivo no debe contener ninguna secuencia de 19 nt con perfecta identidad con otros genes
Otras observaciones científicas recomiendan que los ARNi con dinucleótidos UU en el extremo 3′ son más eficientes. Se pueden diseñar ARNi con otros dinucleótidos salientes. Sin embargo, es preferible evitar los residuos G en el saliente debido a la posibilidad de que la RNasa escinda el siRNA en residuos G monocatenarios.
2. Perfil termodinámico
Algunos estudios has demostrado la importancia del perfil termodinámico del ARNi, que es crucial para la incorporación de ARNi en el RISC. La hebra de ARNi con un extremo 5′ menos termoestable se carga preferiblemente en el RISC, por lo que la baja estabilidad de la hebra antisentido 5′ del ARNi se correlaciona con la eficiencia del ARNi. Una secuencia -AS- con A o U como máximo de 1 a 7 posiciones parece ser más eficiente porque hay menos enlaces de hidrógeno entre A y U que entre C y G da como resultado baja estabilidad.
3. Contenido de GC
Se han descrito diferentes diseños de ARNi donde el %GC varía según el protocolo. Sin embargo, se recomienda que para conseguir mayor eficacia del ARNi, el %GC varíe entre 30-50%, ya que un contenido más alto disminuye la actividad del ARNi
4. Longitud del tallo de ARNi y secuencia del bucle que une las hebras sentido y antisentido de la horquilla de ARNi
La longitud de la secuencia de nucleótidos varía entre los protocolos utilizados, presentando valores de eficacia del ARNi similares entre sí. Como se describe en ThermoFisher, Ambion utiliza secuencias de 19 nucleótidos como base de expresión del ARNi. Otros grupos de investigación utilizan tallos que van desde 21 nucleótidos hasta 25-29 nucleótidos. En la tabla 3 se resume el tamaño del bucle y las secuencias específicas utilizadas.

5. Modificaciones químicas
Muchos estudios demuestran que algunas modificaciones químicas pueden mejorar las propiedades de
ARNi. Las modificaciones pueden aumentar la especificidad y la estabilidad de los ARNi y prolongar el efecto del silenciamiento. Algunas modificaciones químicas tienen poca o ninguna influencia en la eficacia del ARNi, pero otros suprimen el ARNi.
No es posible modificar el extremo 5′ de la secuencia AS, porque las fosforilaciones en el extremo 5′ de la cadena antisentido es esencial para la función del ARNi. Además, se ha visto que el grupo 2-OH de la ribosa no es esencial para el ARNi. Las modificaciones en este grupo químico podrían estabilizar el ARN porque dicho grupo 2-OH, que distingue al ARN del ADN, se requiere para el ataque nucleofílico durante la degradación por RNasas. En la tabla 4 se resumen los efectos de algunas modificaciones químicas en ambas cadenas.
| Cadena antisentido | Cadena molde | Función del ARNi |
| Tapar extremo 5′ | Tapar extremo 3′ | No se afecta |
| Tapar extremo 3′ | Sin modificar | No se afecta |
| Sin modificar | Tapar extremo 5′ | Gravemente afectada |
| Pirimidinas que contienen 2 ́-fluoro ribosa |
Pirimidinas que contienen 2 ́-fluoro ribosa |
No se afecta |
| 2′-desoxirribosa | Sin modificar | Moderadamente afectada |
| Sin modificar | 2′-desoxirribosa | Gravemente afectada |
| 2′-O metil ribosa | Sin modificar | Moderadamente afectada |
| Sin modificar | 2′-O metil ribosa | Gravemente afectada |
| Sin modificar | 2′-O metil ribosa en la posición 2 desde el extremo 5′ | Reduce objetivos no deseados de ARNi |
6. Selección de la secuencia objetivo en el ARNm diana
En la mayoría de los casos, los ARNs interactúan con la región no traducida 3′ (UTR 3′) de los ARNm diana para inducir la degradación del ARNm y la represión traduccional. Sin embargo, también se ha informado la interacción de los ARNs con otras regiones, incluida la 5 ‘UTR, la secuencia codificante y los promotores de genes.
PASO 1. IDENTIFICACIÓN DEL FRAGMENTO DIANA: POLIFENOL OXIDASA
INTRODUCCIÓN
Tal y como se explica en Thermo Fisher, existen varios métodos para preparar siRNA, como la síntesis química, la transcripción in vitro, los vectores de expresión de siRNA y los casetes de expresión de PCR. Independientemente del método que se utilice, para diseñar un siRNA debemos elegir el sitio objetivo del siRNA.
En este trabajo se pretende silenciar el gen de la polifenol oxidasa (PPO, EC 1.14.18.1) del aguacate (persea americana), la cual convierte los fenoles en quinonas, causando el pardeamiento enzimático. La enzima cataliza la O-hidroxilación de monofenoles (fenoles en los cuales el anillo bencénico contiene un único sustituyente hidroxilo) para convertirlos en O-difenoles (fenoles con dos sustituyentes hidroxilo). La misma enzima puede, posteriormente, catalizar la oxidación de los O-difenoles para formar O-quinonas. Las o-quinonas son muy reactivas y atacan a una gran variedad de componentes celulares. La rápida polimerización de las O-quinonas produce pigmentos de color negro, marrón o rojo, lo que a su vez es la causa del pardeamiento enzimático.
Esta enzima se encuentra en organismos procariotas y eucariotas, pero recibe nombres diferentes según el ser vivo del que procede: tirosinasa en animales y procariotas; PPO en vegetales. Algunos estudios han mostrado que, en plantas, dicha enzima se localiza en los cloroplastos. Para su estudio en plantas, se utiliza normalmente como sustrato el ácido clorogénico, un compuesto polifenólico unido a un azúcar, que se puede encontrar de forma natural en diferentes vegetales, incluyendo el aguacate. Para más información sobre la polifenol oxidasa consultar este enlace.
PROCEDIMIENTO
BÚSQUEDA DE LA SECUENCIA DE ADN DEL AGUACATE (Persea americana)
El primer paso para identificar la secuencia diana que se quiere silenciar es la obtención de la secuencia de ADN del aguacate. Para ello, se recurrirá a genes ortólogos que están anotados en NCBI.
Los genes ortólogos son genes homólogos de especies diferentes que evolucionaron a partir de un ancestro común en un proceso de especiación, mientras que los parálogos son resultado de la duplicación.
Nota 1: Es muy importante analizar estas secuencias, ya que no se trata de la secuencia deseada, pero sí nos servirán para acercarnos a dicha secuencia deseada.
- Análisis filogenético
Para la búsqueda de la secuencia de ADN del aguacate debemos preguntarnos cuál o cuáles son las especies más cercanas en términos filogenéticos. Para ello nos ayudaremos de los filtros de NCBI, limitando la ventana de posibilidades a la clase magnolipsida (figura 7)

De esta manera, se obtienen diferentes secuencias del gen que codifica para la PPO en organismos cercanos respecto a la clase filogenética:
- Extremo 3′ del ARNm de la polifenol oxidasa en patata: Aunque NCBI no indica que sea una secuencia truncada, podemos destacar algunas puntuaciones. Por ejemplo, la secuencia peptídica empieza por serina (debería comenzar por metionina). Por otro lado, según el título de la entrada, se trata de la secuencia de un extremo 3′, por lo que probablemente sea una secuencia incompleta. La secuencia del potato tiene un péptido señal (transit peptide). Eso nos indica que se procesará al entrar en el cloroplasto, por lo que la proteína madura (PPO) se obtiene a partir del nucleótido 265. Aunque sea solo la sección 3′-END sabemos que la parte anterior irá cortada, por ser parte de un péptido señal. En conclusión, se puede buscar ortología.
- ARNm de polifenol oxidasa PPO (alelo POT33) de Solanum tuberosum tuber, cds completo: Obtenemos información sobre la existencia de un alelo de dicho gen en la patata que no presenta péptido señal ni está truncado.
- ARNm de polifenol oxidasa de Malus domestica, cds completo
- Gen de L. esculentum para polifenol oxidasa
- Alineamiento múltiple
A continuación, se ha realizado un alineamiento múltiple con Clustal Omega, que mostrará un alineamiento global. En la figura 8 se muestra el esquema filogenético de las secuencias alineadas.

Según la figura 8, las especies que divergieron primero en el momento de evolución fueron la manzana y Solanum tuberosum. Más tarde, la patata y L. esculentum divergieron a la vez.
Este primer alineamiento no aporta mucha información, pero sí puede ser útil para determinar discrepancias o similitudes correctas entre dichas secuencias. Es decir, permite comprobar si es cierto que existe un parecido significativo entre ellas debido a la evolución, que nos permita acceder a la secuencia de persea americana (organismo deseado). Además, este alineamiento permitirá obtener cebadores degenerados.
- Búsqueda de una secuencia similar
Para buscar una secuencia similar a las secuencias mencionadas en el apartado anterior, en concreto en el organismo persea americana, se utilizará la base de datos NCBI, siguiendo los siguientes pasos.
Antes de dar comienzo a los pasos para la búsqueda de la secuencia similar, hay que destacar algunas bases de datos que NCBI utiliza para realizar el alineamiento (figura 9)

Nucleotide collection se creó cuando se formó GenBank a principios de 1980. Los primeros intentos de secuenciación del genoma se realizaron con Sanger, por lo que en esta base de datos encontraremos fundamentalmente secuencias largas secuenciadas con Sanger.
En RefSeq se encuentran secuencias de referencia. Todo el mundo tiene acceso para mandar sus trabajos, pero algunas secuencias tienen índices de calidad mejores que otros (por ejemplo, indicando la redundancia al secuenciar varias veces). Otro concepto de calidad es la medicion de la actividad enzimatica (GenMak o August son programas que predicen dónde hay intrones, exones, etc. de genomas enteros y, por similaridad con otros ortólogos, deduce la posible actividad biológica de una proteína).
Cuando se secuencia un genoma, en principio hay que romper el genoma y luego ensamblarlo para poder secuenciarlo. Shotgun es una tecnología que hace posible romper el genoma de ADN para posteriormente, secuenciarlo. Una vez tienes el fragmento de ADN, hay que generar la continuidad de la molecula para volver a generar el genoma. Existe una base de datos (wtg) del genoma “en su etapa inicial” que consiste en anotar dichos fragmentos rotos antes de su secuenciación. Se diferencian con los anteriores en que no se sabe dónde están los genes ni cuántos exones o intrones hay. Sin embargo, permite encontrar una secuencia de un gen, como el de la polifenol oxidasa del aguacate.
1. Secuencia similar al extremo 3′ del ARNm de la polifenol oxidasa en patata
Empezaré buscando una secuencia similar a la de la patata. Para ello, se selecciona la región 2…1768 de la patata, porque como indica NCBI, esta es la región codificante (CDS) de la polifenol oxidasa en este organismo.
Se encuentran 6 secuencias con un alineamiento significativo, de las cuales la primera secuencia presenta un E-value razonablemente pequeño como para pensar que este alineamiento no se debe al azar. Además, presenta un alto porcentaje de identidad (78,38%), teniendo en cuenta las diferencias entre especies. Sin embargo, cabe destacar que el porcentaje de cobertura de las secuencias alineadas es muy bajo, lo que indica que ha encontrado dichos porcentajes de identidad con regiones muy cortas (de algunos nucleótidos) con la secuencia deseada (figura 10).

2. Secuencia similar a ARNm de polifenol oxidasa PPO (alelo POT33) de Solanum tuberosum tuber, cds completo
Para ello, se selecciona la región 11…1810 de Solanum tuberosum, porque como indica NCBI, esta es la región codificante (CDS) de la polifenol oxidasa en este organismo.
Se encuentran 11 secuencias en persea americana con un alineamiento significativo, de las cuales la cuarta secuencia presenta un E-value razonablemente pequeño como para pensar que este alineamiento no se debe al azar. Además, presenta un alto porcentaje de identidad (78,21%), teniendo en cuenta las diferencias entre especies. Sin embargo, al igual que ocurría con la secuencia similar a la patata anterior, los porcentajes de cobertura de las secuencias alineadas son demasiado bajos (figura 11).

3. Secuencia similar al ARNm de polifenol oxidasa de Malus domestica, cds completo
Continuaré buscando una secuencia similar a la de Malus domestica del apartado anterior. Para ello, se selecciona la región 19…1800 de Malus domestica, porque como indica NCBI, esta es la región codificante (CDS) de la polifenol oxidasa en este organismo.
Se encuentran 10 secuencias en persea americana con un alineamiento significativo, de las cuales la primera secuencia presenta un E-value razonablemente pequeño como para pensar que este alineamiento no se debe al azar. Además, presenta un alto porcentaje de identidad (71,08%), teniendo en cuenta las diferencias entre especies (figura 12).

4. Secuencia similar al gen de L. esculentum para polifenol oxidasa
A continuación, se selecciona la región 1208…2965 de L. esculentum, porque como indica NCBI, esta es la región codificante (CDS) de la polifenol oxidasa en este organismo.
Se encuentran 7 secuencias en persea americana con un alineamiento significativo, de las cuales ninguna secuencia presenta un E-value razonablemente pequeño, si se comparan con los anteriores valores de E-value obtenidos. Presenta un alto porcentaje de identidad 67,88%), teniendo en cuenta las diferencias entre especies (figura 13).

Teniendo en cuenta los resultados obtenidos, el alineamiento encontrado con Malus domestica es el que mejor relación de porcentajes (E-value, porcentaje de cobertura, porcentaje de identidad, etc.) tiene con persea americana. Se concluye que la secuencia con número de acceso SDXN01000094.1 es la secuencia más probable de la polifenol oxidasa de persea americana.
- Comprobación de la actividad polifenol oxidasa de la secuencia nucleotídica
El alineamiento de secuencias nucleotídicas nos ha acercado a una secuencia probable del gen que codifica la polifenol oxidasa en persea americana (figura 14). Sin embargo, en esta sección se discutirá alguna evidencia más de su actividad como enzima deseada.

Para ello se realizará un BLAST X que nos proporcione un alineamiento de la secuencia peptídica qu se obtiene de la secuencia nucleotídica alineada anteriormente (925462…926198). En la figura 15 se observa que la primera secuencia proteíca presenta un E-value igual a 0 y un porcentaje de identidad igual al 98,78% tras un porcentaje de cobertura de casi el 100% (99% en concreto). Esto nos indica que la proteína codificada de dicha región presenta una alta similitud de identidad con una proteína que presenta actividad polifenol oxidasa.

Se concluye así que hemos encontrado la secuencia que codifica la polifenol oxidasa en persea americana (SDXN01000094.1).
- Información adicional del BLAST X y búsqueda de posibles intrones
Tras el BLAST X se consigue un alineamiento protéico entre la región que posiblemente codifica la polifenol oxidasa en persea americana y la región que codifica la polifenol oxidasa en Cinnamomum micranthum (figura 16).
Nota 2. Cabe resaltar que se trata de una suposición indirecta, porque se está asignando dicha actividad enzimática a una proteína por el parecido y la similitud con otra proteína que sí presenta esta actividad. Al tratarse de una suposición indirecta no hay que olvidar nunca que cabe cierta probabilidad de error.

Este alineamiento comienza por el aminoácido 228 (subject), lo que nos indica que en Cinnamomum micranthum faltan 227 aminoácidos del principio por alinear, como mínimo. Además, la longitud de la secuencia protéica de Cinnamomum micranthum es de 640 aminoácidos, pero el alineamiento acaba en el aminoácido número 472, lo que indica que también faltan 168 aminoácidos del final. Por otro lado, la secuencia de nucleótidos no empieza por metionina (el codón de inicio AUG codifica metionina). Esta información se puede interpretar como la posibilidad de una secuencia truncada.
Teniendo en cuenta que ambas secuencias protéicas alineadas están en sentidos opuestos, se ampliará la ventana de nucleótidos en la secuencia de persea americana de la siguiente forma, con el objetivo de buscar las regiones iniciales y finales del gen:
– Al nucleótido número 926198 se suman 1050 nucleótidos
– Al nucleótido número 925462 se restan 1300 nucleótidos
Otra consecuencia de la similitud existente entre secuencias alineadas (puntos anteriores) es la siguiente afirmación: Se puede pensar que si una secuencia nucleotídica (incluso peptídica) se ha conservado a lo largo de la evolución con tan alto porcentaje de identidad, la estructura génica, la existencia de intrones y exones también se habrá conservado con un alto porcentaje de probabilidad. Por lo tanto, si las secuencias que codifican PPO en organismos como Malus malusca, Cinnamomum micranthum, patata, etc., no presentan intrones, es probable que tampoco estén presentes en persea americana.
Tras realizar un alineamiento con la posible región inicial del gen (nucleótidos 926198 hasta 927248) observamos que el nucleótido número 927218 en persea americana alinea con región inicial de la secuencia de Cinnamomum micranthum. Esto indica que alrededor de dicho nucleótido en persea americana se encontrará la zona inicial del gen (5’UTR) (figura 17)

Tras realizar un alineamiento con la posible región final del gen (nucleótidos 925464 hasta 926964) observamos que el nucleótido número 924175 en persea americana, alinea con la zona final de la secuencia de Cinnamomum micranthum. Esto indica que alrededor de dicho nucleótido en persea americana se encontrará la zona final del gen (3’UTR) (figura 18)

Con la base de datos EXPASY se ha obtenido el marco de lectura de la secuencia de nucleótidos en persea americana, para corroborar si el codón stop concuerda con el alineamiento de la figura 18
PASO 2. DISEÑO DE CEBADORES
INTRODUCCIÓN
- Rango que se va a amplificar
Como se explicaba en las consideraciones iniciales para el diseño del iRNA, la interacción entre los ARNs (tales como el iRNA) y los ARNm se ve que ocurre en la región 3’UTR con alta probabilidad y eficiencia. De esta manera, se amplificará la región 3’UTR del gen que codifica a la PPO en persea americana.
Según la base de datos NCBI, la región 3’UTR de la secuencia de persea americana se puede observar en la figura . Además, teniendo en cuenta que una longitud óptima de DNA plantilla para el dsRNAs es más de 400 pb, el intervalo de nucleótidos será el siguiente:
– 924175 … 924375 (200 pb, para que al final se obtenga un dsRNA de 400 pb o más)
Por tanto, se diseñarán 2 cebadores teniendo en cuenta dicho rango.
- Afinidad por otros sitios de unión
Hay que tener en cuenta que los cebadores pueden tener afinidad por otros sitios de unión dentro de la secuencia que queremos amplificar. Por lo tanto, si los cebadores diseñados presentan esta característica, vamos a priorizar aquellos que tengan más errores en el extremo 3′ que en el 5′ de la secuencia.
Esto se debe a que la Taq polimerasa se encarga de amplificar la secuencia de nucleótidos desde un extremo 3′. Por lo tanto, cuando encuentre un extremo 3′ erróneo se hará más complicada su unión y, por lo tanto, su amplificación. De esta manera, se evita la amplificación de extremos no deseados.
- Temperatura de fusión (Tm)
La temperatura de fusión (Tm) es la temperatura en la cual el 50% del ADN tiene sus hebras separadas (desnaturalización del ADN).
La temperatura de fusión condiciona el diseño de cebadores, así como la diferencia entre la Tm de los cebadores y la Tm del ADN. Con una diferencia de Tm entre 15-20º conseguiremos que el cebador compita con el producto (ADN) y no se pegue el producto entre sí.
Por lo tanto, antes de comenzar con el diseño de cebadores, tenemos que calcular la Tm del ADN.
CÁLCULO DE LA Tm DE ADN
Existen algunas páginas web que calculan la Tm del ADN, como ENDMEMO, EMBOSS, etc. Sin embargo, para la mayoría es necesario conocer la concentración del ADN problema. Nanodrop es un dispositivo capaz de medir pequeños volúmenes de ADN, ARN o proteínas, y sirve para cuantificar de manera exacta y fiable muestras que no son fácilmente medibles con otros utensilios (vídeo 3).
En ThermoFisher Scientific podemos encontrar diferentes dispositivos para la cuantificación de ARN/ADN a través de espectroscopía UV-visible, los cuales incluyen el espectrofotómetro NanoDrop One para un análisis práctico de microvolúmenes de una sola muestra (nº de catálogo ND-ONE-W) por 10.850€. Este dispositivo permite el uso de concentraciones bajas 2,0 ng/µl de ADNdc.
Algunas bibliotecas estándar de Illumina, como Nextera, requieren el uso de métodos de tinción fluorescente específicos de dsDNA para una cuantificación precisa. Estos métodos suelen medir la concentración de dsDNA en ng/µl. Para convertir de ng/µl a nM puede seguirse la siguiente ecuación:

Asumiendo que Nanodrop mide 2 nanogramos y que el protocolo de PCR qur utilizaremos para amplificar la secuencia se realiza en un volumen de 25 µl. Además, el rango que se va a amplificar tiene 200 pb:
(2 ng/ 25 µl) / (660 g/mol x 200 pb) x 106= 0,606 nM es la concentración de ADN plantilla
Una vez conocida la concentración de ADN, podemos calcular la Tm del ADN plantilla a través de EMBOSS, la cual necesita los datos de la concentración de iones (50mM de K+) que se utilizará en el proceso de amplificación del ADN por PCR:
– Tm del ADN plantilla: 86,6 ºC
Teniendo en cuenta que EMBOSS aumenta alrededor de 15º la Tm:
– Tm del ADN plantilla: 71,6 ºC
Una vez conocida la Tm del ADN, podemos determinar la Tm de los cebadores con la consideración anteriormente mencionada: Tm ADN – Tm cebador más inestable = 15-20º
– Tm para cebadores: (68,8 – 15) = 56,6 ºC
Es importante mencionar que para estos cálculos se ha utilizado como concentración de sal la que corresponde a la sal KCl del tampón de PCR, en lugar de la concentración de sal MgCl2. Esto se debe principalmente a la despreciable cantidad de sal Mg2+, a la importancia de la sal K+ (ver enlace), así como a la despreciable cantidad de sal K+ dentro de la Taq DNA polimerasa
PROCEDIMIENTO
Damos paso al procedimiento para el diseño y selección de los cebadores que usaremos para amplificar el ADN, a través de la base de datos NCBI (pick primers), como se observa en la figura 19.
 |
 |
Con las restricciones de la figura 19 y los datos mencionados, obtenemos los siguientes cebadores (Figura 20).

Con la mayoría de cebadores obtenidos se formará un producto de amplificación de alrededor 320 pb. Tras los tratamientos posteriores que conlleva la síntesis de la doble cadena de ARN, se formará una molécula de unos 700 pb. Teniendo en cuenta que, según la bibliografía consultada, el ADN plantilla para la síntesis de la doble cadena de ARN debe ser de más de 400 pb, dicha longitud final no supondrá un problema. Esta doble cadena de ARN poseriormente será procesada por los componentes de la planta, para dar lugar al iRNA.
Otro parámetro a tener en cuenta es la autocomplementariedad de los cebadores en el extremo 3′, lo cual indicará la probabilidad de que ambos cebadores formen dímeros entre sí, disminuyendo la eficacia de la reacción. Además, se deben escoger parejas de cebadores que presenten valores de Tm cercanos entre sí, y que estén cerca de la Tm óptima (56,6 ºC).
Por lo tanto, la pareja de cebadores nº 7 presenta mayores ventajas que el resto (figura 21).

A la secuencia de cada cebador se le añadirá una secuencia diana de dos enzimas de restricción (BamHI-HF y XhoI). Las secuencias diana y los sitios de corte de las enzimas de restricción BamHI-HF y XhoI son las siguientes:
BamHI-HF:
5′ G^GATCC 3′
3′ CCTAG^G 5′
XhoI:
5′ C^TCGAG 3′
3′ GAGCT^C 5′
Por otro lado, la actividad y eficiencia en el corte de las enzimas de restricción se ve alterada al añadir pares de bases a su secuencia diana, pudiendo darse el caso de que la enzima no llegara a actuar si no se añaden algunas pares de bases. En este caso, la eficiencia del corte de las dos enzimas de restricción de interés al añadir algunas pares de bases se observan en la tabla 5.
| Enzima | 1 pb desde el final | 2 pb desde el final | 3 pb desde el final | 4 pb desde el final | 5 pb desde el final |
| BamHI-HF | + | + | +++ | +++ | +++ |
| XhoI | ++ | ++ | ++ | +++ | +++ |
De esta forma, las secuencias diana de la enzima de restricción BamHI-HF se añadirá al extremo 5′ del cebador forward, adicionando 3 pb al final de su secuencia, obteniendo la siguiente secuencia de cebador:
5′ GGGGGATCCAATTCTGGCAAACAGAGGC 3′
La secuencia diana de la enzima de restricción XhoI se añadirá al extremo 5′ del cebador reverse, adicionando 4 pb al final de su secuencia, obteniendo la siguiente secuencia de cebador:
5′ GGGGCTCGAGTCTGTTGGAGGGTTGAAGATA 3′
PEDIDO DE CEBADORES
Una vez hemos diseñado y seleccionado los 2 cebadores tenemos que realizar el pedido. En este caso, se encargarán los cebadores a ThermoFisher Scientific (tabla 6)
| Forward | Reverse | Total | |
| Precio (€) | 5,40 | 5,22 | 10,62 |
| Cantidad garantizada (nmol) | 6,0 | 6,2 | 12,2 |
| Cantidad pedida (nmol) | 25 | 25 | 50 |
PASO 3. AMPLIFICACIÓN DEL ADN PLANTILLA: polymerase chain reaction (PCR)
INTRODUCCIÓN
Antes de comenzar con el procedimiento de la PCR, existen algunas consideraciones y precauciones que debemos tener en cuenta. Estas se pueden resumir en el vídeo 4.
A pesar de que el vídeo 4 explica un protocolo para realizar la PCR, en este caso se utilizará Thermo Scientific Taq DNA Polymerase with KCl Buffer, ya que, a diferencia del protocolo del vídeo 4, Thermo Scientific Taq DNA polymerase utiliza KCl para la reacción de PCR. Es importante tener en cuenta la sal de KCl en el tampón de PCR (figura materiales) porque actúa neutralizando la carga presente en el esqueleto del ADN. Para más información sobre la importancia del KCl en la reacción de PCR, así como la presencia de MgCl2, puede consultarse el siguiente enlace.
La información sobre pedidos puede hacerse siguiendo este enlace, o bien, buscando a través de los códigos aportados por ThermoFisher (tabla 7).

MATERIALES
- Componentes suministrados por el kit ThermoScientific Taq DNA Polymerase with KCl Buffer se muestran en la tabla 8
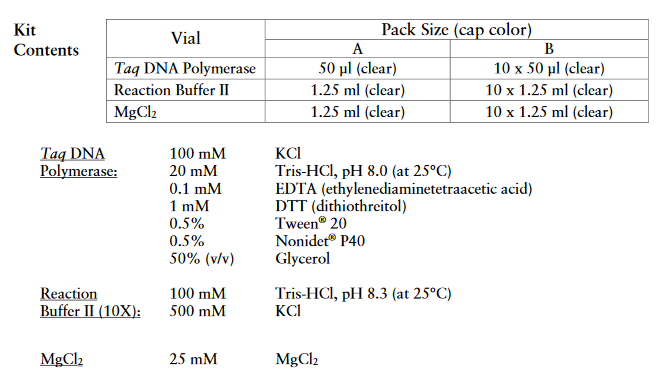
- Componentes no suminstrados por el kit
– dNTP Mix (20mM): Comprar aquí un set de dNTP (nº de catálogo R0186) por 1.414,00€
– Agua de calidad PCR: Comprar aquí (nº de catálogo W1754-5VL) por 182,00€
– Termociclador: Comprar aquí (nº de catálogo 15806731) por 9698.36€
PROCEDIMIENTO
CÁLCULOS PREVIOS
Antes de comenzar con los pasos descritos en el protocolo para amplificar el ADN, debemos manipular y diluir los cebadores para su uso en el PCR. Tenemos que considerar las concentraciones y volúmenes descritos en el protocolo, mostrados en la tabla 9.

- ¿Qué concentración de cebadores hay que preparar?
Para calcular la concentración de solución madre de cebadores que debemos preparar hay que tener en cuenta los siguientes datos:
- Se añaden 1,25 µl (según tabla 9)
- La concentración final de cada cebador es de 0,5 µM (según tabla 9)
- El tubo de PCR es de 25 µl (según tabla 9)
Por lo tanto, la cantidad de cebadores que hay que añadir en 25 µl, para que la concentración de cebadores sea de 0,5 µM es:
1 L = 106 µl –> 0,5 µmol
25 µl –> x µmol
x = 0,0000125 µmol = 12,5 pmol
Estos 12,5 pmol tenemos que añadirlos a 1,25 µl y obtendremos la siguiente concentración de cebadores:
1 L = 106 µl –> x pM
1,25 µl –>12,5 pmol
x = 10000000 pM = 10 µM
- ¿Cómo preparo 10 µM de cebadores?
Aquí debemos considerar que, muchas veces, las empresas nos proporcionan una cantidad de cebadores que no se corresponde a la que ellos nos indican. Por eso, sería necesaria una cuantificación previa utilizando Nanodrop.
Para preparar una concentración de 10 µM de cada cebador hay que tener en cuenta los siguientes datos:
- Tenemos 25 nmol de cebadores (según tabla 6)
- Los tubos albergan como máximo 1,5 ml
- Queremos preparar 10 µM de cada cebador (en total hay que preparar 2 cebadores)
10 µmol = 104 nmol –> 103 ml
25 nmol –> x ml
x = 2,5 ml
Los tubos no pueden albergar 2,5 ml por lo que tendríamos que hacer la siguiente consideración: En lugar de echar 2,5 ml (2500 µl) de agua al tubo, añadiremos 0,25 ml (250 µl), de manera que obtendremos un tubo 10 veces más concentrado. Podemos comprobarlo de la siguiente manera:
250 µl –> 25 nmol
106 µl –> 100000 nM = 100 µM
Para solucionar el aumento de concentración en el tubo, podemos diluir 10 veces añadiendo 10μL de
solución madre a 90μL de tampón (obtendríamos así 10μM). Esto lo podemos distribuir en un total de 10 tubos (dilución 10+90). Así, si se contamina algún tubo o cometemos algún error, tenemos margen para deshechar algún tubo.
- ¿Cuántas reacciones de PCR se pueden llegar a realizar?
- Tubo de PCR de 25 µl (según tabla 9)
- La concentración final de cada cebador es de 0,5 µM (según tabla 9)
- Hay que añadir 12,5 pmol de cada cebador en los tubos (según cálculos anteriores)
25 nmol = 25000 pmol en total
25000 pmol en total / 12,5 pmol en cada tubo = 2000 reacciones posibles
PROTOCOLO DE PCR
Una vez realizados los cálculos, podemos seguir los pasos que se describen en el protocolo de PCR (figura 22).

RECOMENDACIONES
– Usar un gradiente de temperatura de los tubos en la primera reacción de PCR , para poder seleccionar aquella temperatura con la que se hayan obtenido mejores resultados.
– Utilizar puntas de pipetas esterilizadas con radiación gamma, así como pinzas, guantes y gorros esterilizados, para no contaminar las muestras. En este enlace se pueden conseguir puntas desechables esterilizadas con filtro.
– Mantener en hielo los reactivos de la PCR, así como los pocillos.
– Para más información sobre cómo hacer un protocolo básico de PCR, así como algunas consideraciones y errores comunes, pinchar este enlace.
– Utilizaremos TE preferentemente en los procesos de extracción de ADN, dilución de cebadores, reacción de PCR, etc. Este tampón no contiene acetato ni borato, por lo que los procesos enzimáticos posteriores no se verán inhibidos.
PASO 4. PURIFICACIÓN DEL ADN: ELECTROFORESIS
INTRODUCCIÓN
El fundamento teórico y las aplicaciones de la electroforesis en gel de agarosa pueden visualizarse en el vídeo 5.
Además, para una descripción más detallada sobre la electroforesis y el gel de agarosa se puede consultar el siguiente enlace, debido a que no es el objetivo de este estudio.
MATERIALES
En este enlace podemos encontrar las tablas de materiales necesarios para realizar una electroforesis en gel de agarosa, así como las cantidades recomendadas según el tamaño de ADN.
Antes de describir los materiales y cantidades necesarias, debemos realizar la elección del porcentaje de gel de agarosa, según el tamaño del fragmento de ADN, ya que esto determinará al resto de componentes (Tabla 10).

Se utilizará un pocentaje de 1,5% para cada electroforesis.
Se utilizará el kit de reactivos (ELECKIT) proporcionados por Danagen, el cual se puede conseguir en el siguiente enlace.
- Componentes proporcionados por el kit ELECKIT
Los materiales proporcionados por ELECKIT se pueden consultar en la tabla 11.

- Componentes no proporcionados por ELECKIT
– Cubetas y fuentes de electroforesis: Conseguir aquí (consultar el precio con Danagen)
– Marcadores de peso molecular: Conseguir aquí (consultar el precio con Danagen). Se comprarán teniendo en cuenta el tamaño de nuestra muestra
PROCEDIMIENTO
1. Añadir la pastilla de DANAGAROSE TABLET (según tabla 12) al volumen necesario de TAE 1X y esperar al menos 2 minutos antes de calentar.

2. Calentar en el microondas a elevada potencia , fundir la agarosa permitiendo una ebullición de 30 segundos aproximadamente.
3. Mezclar bien.
4. Volver a calentar en el microondas hasta que la solución esté transparente.
5. Enfriar la solución a unos 55ºC. A esta temperatura añadir el GELSAFE Nucleic Acid Stain*.
6. Añadir unos 2.5 microlitros de GELSAFE Nucleic Acid Stain por cada 50 ml de gel de agarosa.
7. Permitir que el gel de agarosa se enfríe hasta que se solidifique.
8. Cargar las muestras utilizando el tampón de carga suministrado 6X DNA Loading buffer y realizar la electroforesis.
9. Detectar las bandas bajo iluminación UV
Según el protocolo, es posible que precipite los componentes de este producto debido a su elevada
concentración. Resuspender con vortex o mediante micopipeta cuando haya pasado mucho tiempo desde su último o uso. Además, los filtros rojo/naranjas utilizados para el bromuro de etidio no deben usarse con el GELSAFE, deben utilizarse los mismos filtros que se utilizan con el SYBR Green. También pueden utilizarse filtros amarillos, verdes o filtros de celofán
También se recomienda ejecutar los geles a un voltaje bajo y constante (~ 10 V/cm) para minimizar los efectos de calentamiento actuales y asimétricos, que pueden inducir artefactos de banda y resolución deficiente.
Hay que tener en cuenta que no veremos las bandas de cebadores: Son fragmentos muy pequeños y no emiten fluorescencia. Es tan pequeña que se han quedado en el tampón.
PASO 5. SISTEMA DE CLONACIÓN: SISTEMA GATEWAY
INTRODUCCIÓN
La clonación molecular se utiliza para amplificar y manipular genes de interés y, posteriormente, insertarlos en plásmidos para así conseguir la replicación y expresión de proteínas. Se conoce como vector de expresión o construcción de expresión a aquellos vectores que se utilizan para la expresión de diferentes genes de interés manipulados o no, es decir, para la síntesis de proteínas.
El vector es un portador, una molécula de ADN de doble cadena cuya misión es unirse con el
fragmento de ADN que se quiere clonar para facilitar su entrada en la célula anfitriona y su
replicación. Es decir, es un vehículo para introducir el gen en la célula anfitriona y es capaz de
replicarse y producir un gran número de copas dentro de la célula anfitriona. Debe tener una serie
de características:
- Capacidad de replicarse autónomamente. Deben poseer orígenes de replicación
independientes de los de la célula hospedadora, y además han de ser de control relajado
para dar lugar a un gran número de copias. - Contener sitios únicos para el reconocimiento de distintas enzimas de restricción. La mayoría de los vectores tienen un sitio de clonación múltiple (MCS), un fragmento de ADN que contiene una serie de sitios únicos de restricción, muy cercanos entre sí con una amplia gama de posibilidades de insertar cualquier fragmento de ADN.
- Portar marcadores de selección que confieran alguna característica fenotípica que permita distinguir las células hospedadoras que los han incorporado de aquellas que no contienen vector. Generalmente los marcadores de selección son genes que confieren resistencia a antibióticos o genes que codifican para productos ausentes en la célula hospedadora
- Ser fácilmente aislables a partir de la célula hospedadora, por lo cual es conveniente que sean de pequeño tamaño, ya que se manipulan con mayor facilidad fuera del sistema celular que las moléculas de DNA grandes que tienden a ser más frágiles.
En esta sección se mencionarán algunos métodos de clonación populares utilizados para crear ADN recombinante (para más información consultar el si siguiente enlace). Sin embargo, se profundizará en el método de clonación GateWay, porque es el sistema más apropiado para el objetivo de esta sección.
CLONACIÓN DE ENZIMAS DE RESTRICCIÓN
Se basa en la capacidad que tienen algunas enzimas de restricción, producidas por arqueas o bacterias, de escindir el ADN de doble cadena en fragmentos de interés, generando extremos romos (extremos sin salientres) o extremos cohesivos (contienen salientes monocatenarios 5′ o 3′). Los extremos protuberantes o romos de diferentes moléculas de dsDNA, cortados con enzimas de restricción, pueden unirse mediante complementariedad de bases por una DNA ligasa, creando así moléculas de ADN recombinante (figura 23).
- Ventajas:
– Cientos de enzimas disponibles
– Baratas
– Cortan secuencias diana específicas
– Producen extremos resultantes predecibles
– Los plásmidos contienen varias secuencias diana para las enzimas de restricción
- Inconvenientes:
– Si se repiten las secuencias diana para las enzimas de restricción, se puede dificultar el trabajo de clonación
– Se quedanpequeñas cicatrices en la secuencia de ADN tras el tratamiento con enzimas de restricción

ENSAMBLAJE DE GIBSON
Utilizando las 3 actividades enzimáticas más comunes en biología molecular (5′ exonucleasa, polimerasa y ligasa), se consigue generar salientes monocatenarios 3′, que luego se fusionan formando una pieza de ADN de doble cadena, en una reacción isotérmica. La actividad 5′ exonucleasa digiere extremos 5′, que luego serán rellenados por la actividad polimerasa y sellados por la ligasa, según el siguiente esquema (figura 24).
- Ventajas:
– Permite ensamblaje simple de múltiples fragmentos de ADN en la orientación deseada
– No necesita secuencias no deseadas en las uniones (sitios diana de enzimas de restricción)
– Se puede usar cualquier fragmento de ADN de doble cadena
- Inconvenientes:
– Funciona mejor con la fusión de 200 pb (fragmentos de menor tamaño son susceptibles de la digestión completa de la exonucleasa).

SISTEMA GATEWAY
La clonación Gateway se basa en la recombinación. Para ello, se necesitan sitios de recombinación específicos, vectores donadores, de entrada y de destino, y las enzimas que van a catalizar estas reacciones.
- Reacción combinada BP clonasa / LR clonasa
En primer lugar, la reacción BP clonasa consiste en la generación de un clon de entrada. Para ello, se parte de un fragmento de ADN (gen de interés) que se desea clonar en un plásmido (plásmido donante). Dicho gen de interés debe estar flanqueado por los sitios de recombinación específicos (attB1 y attB2, se puede conseguir, por ejemplo, mediante reacción de PCR), y el plásmido donante debe estar flanqueado por sitios attP compatibles. Tras la acción de la BP clonasa se obtiene un clon de entrada con sitios attL recombinados que flanquean al gen de interés (figura 25). El uso de vectores TOPO y vectores de enzimas de restricción/amplificación por PCR son las formas más comunes de construir su propio clon entrada.

Posteriormente, el gen de interés puede transferirse rápidamente a cualquier vector Gateway® Destination compatible que contiene sitios attR a través de las enzimas LR clonasa (figura 26).

- Reacción LR clonasa individual
Se puede simplificar el sistema Gateway omitiendo la reacción de BP clonasa. En este sentido, se parte de un gen de interés flanqueado por secuencias diana de enzimas de restricción que también tengan su secuencia diana entre los sitios attL del plásmido de entrada. De esta forma, las enzimas de restricción que originen extremos cohesivos, permitirán la recombinación entre el gen de interés y el plásmido de entrada.
Otra alternativa para insertar un gen de interés en el plásmido de entrada, sin la necesidad de utilizar enzimas de restricción, es utilizar un plásmido de entrada TOPO (vector pENTR/TOPO). Para ello, se direcciona el producto de PCR al sitio donde la topoisomerasa I actúa en el plásmido (entre attL1 y attL2). La misma topoisomerasa actúa escindiendo el enlace fosfodiéster en una cadena y, cuando se une un fragmento de interés complementario, fusiona el ADN en el extremo 3′
Posteriormente, el vector de entrada (pENTR o pENTR/TOPO) puede someterse a una reacción catalizada por la LR clonasa, haciendo posible la recombinación entre un vector de entrada con sitios attL y un vector de destino con sitios attR (figura 26).
Nota 3: En este trabajo no se profundizará más acerca de los vectores de entrada TOPO (pENTR/TOPO), debido a que se va a trabajar con un vector de entrada pENTR y enzimas de restricción (tabla 5)
MATERIALES
Una vez introducido el fundamento teórico de este sistema de clonación, así como algunas alternativas, se procede a describir los componentes que se va a utilizar:
PARTE UNO: INTRODUCCIÓN DEL FRAGMENTO DE ADN EN EL VECTOR DE ENTRADA
- Vector de entrada
Se utilizará un vector de entrada proporcionado por ThermoFisher llamado Gateway™ pENTR™ 2B Dual Selection Vector (figura 27). Se puede comprar aquí (nº de catálogo A10463) por 57,25€.
Las características que ofrece este vector de entrada se presentan a continuación:
- Sitios attL1 (bases 358-457) y attL2 (bases 1984-2083) para la recombinación específica del clon de entrada con un vector de destino Gateway™ para garantizar la clonación del gen de interés en la orientación correcta para su expresión
- Secuencia de consenso de Kozak para la iniciación eficiente de la conversión en sistemas eucariotas
- Secuencias de terminación de la transcripción rrnB para evitar la expresión basal del producto PCR de interés en E. coli
- Origen de pUC para la elevada replicación de copias y el mantenimiento del plásmido en E. coli
- Gen de resistencia a la kanamicina para su selección en E. coli
- El gen de fusión de ccdB⁄cloranfenicol ubicado entre los dos sitios attL para:
– selección negativa – selección de cloranfenicol en E. coli - Gen de resistencia a la kanamicina para su selección en E. coli

- Enzimas de restricción
En la figura 27 se observan las enzimas de restricción que tienen añguna región diana entre los sitios attL1 y attL2.
Para la elección de la enzima de restricción que se va a utilizar, debe tenerse en cuenta los siguientes puntos:
- La enzima de restricción genera extremos cohesivos, para la posterior recombinación de extremos.
- La enzima de restricción no tiene más de un sitio de corte en zonas indeseadas en el plásmido, porque esto podría complicar la introducción del inserto en la zona deseada
- La enzima de restricción no tiene sitio de corte en el interior del fragmento de ADN que se desea insertar
Se han escogido las enzimas de restricción BamHI y XhoI (tabla 13), para la inserción del fragmento de ADN en el vector de entrada. Dicha inserción requiere que se generen extremos cohesivos complementarios al plásmido en el fragmento de ADN. Para ello, se han diseñado cebadores que tienen la secuencia diana de estas enzimas de restricción (PASO 2. DISEÑO DE CEBADORES)
| BamHI-HF | XhoI | |
| nº catálogo | R3136 | R0146 |
| Precio (€) | 63,00 | 74,00 |
| Concentración (units/ml) | 20,000 | 20,000 |
| Temperatura (ºC) | 37 | 37 |
| Actividad en buffer NEB (rCutSmart) (%) | 100 | 100 |
| Actividad en buffer NEB (r1.1) (%) | 100 | 75 |
| Actividad en buffer NEB (r2.1) (%) | 50 | 100 |
| Actividad en buffer NEB (r3.1) (%) | 10 | 100 |
| Inactivación por calor (ºC) | No | 65 |
| Secuencia diana |  |
 |
| Para más información | Consultar link | Consultar link |
Nota 4: (1) Digerir con rCutSmart Buffer (o NEBuffer 4 + rAlbumin) a 37 °C. (2) BamHI-HF tiene un isosquizómero (BamHI) que presenta más porcentaje de actividad en el buffer NEB r3.1. Sin embargo, este no será utilizado, porque presenta una actividad estrella (star activity) en la mayoría de buffer utilizados. BamHI-HF es una enzima modificada que tiene una actividad estrella significativamente reducida
Para la doble digestión del fragmento de ADN y del plásmido mediante enzimas de resitricción, también se utilizarán los siguientes componentes. Pueden conseguirse a través de BioLabs.
– 10X rCutSmart Buffer: Comprar aquí (nº catálogo B6004S) por 27,02€. Al utilizar este buffer se está teniendo en cuenta que ambas enzimas presentan un porcentaje de actividad elevado y, además, no presentan actividad estrella bajo las condiciones de este buffer (tabla 10)
– Nuclease-free Water: Comprar aquí (nº catálogo B1500L). Precio no disponible
– Incubadora de laboratorio: Utilizar la que se compró para el protocolo de la determinación de la concentración de proteínas.
Además, para la reacción de ligación se necesitarán los siguientes componentes:
– T4 DNA ligasa: Comprar aquí (nº catálogo M0202S). Precio no disponible
- Células competentes
Para la preparación de células competentes se utilizarán son las recomendadas por el manual del vector de entrada (figura 26). Estas son las células TOP10. Comprar aquí E. coli químicamente competente One Shot™ TOP10 (nº catálogo C404010) por 300,00€.
Las células de E. coli de One Shot™ TOP10 presenta las siguientes características:
- hsdR para una transformación eficaz de ADN no metilado procedente de amplificaciones de PCR
- mcrA para una transformación eficaz de ADN metilado procedente de preparaciones genómicas
- lacZΔM15 para la detección de color blanco o azul de clones recombinantes
- endA1 para preparaciones de ADN de limpiador y una mejora de los resultados en las aplicaciones secuencia abajo gracias a la eliminación de la digestión no específica por parte de Endonuclease I
- recA1 para reducir los casos de recombinación no específica en ADN clonado
- Genotipo: F- mcrA Δ(mrr-hsdRMS-mcrBC) Φ80lacZΔM15 Δ lacX74 recA1 araD139 Δ(araleu)7697 galU galK rpsL (StrR) endA1 nupG
- Eficacia de transformación de 1 x 109 ufc/µg de ADN plasmídico que resultan ideales para la clonación y la propagación plasmídica de alta eficacia.
Nota 5: La eficiencia de transformación de las células competentes es una consideración importante. Los fabricantes proporcionan la eficiencia de transformación de las células competentes en unidades formadoras de colonias por microgramo de ADN (UFC/µg), generalmente en un rango de 1 x 106 a 1 x 109 UFC/µg. En estrategias de clonación y ligadura más difíciles, la elección de células con mayores eficiencias de transformación pueden mejorar en gran medida la probabilidad de obtener clones deseados.
Los componentes suministrados por E. coli químicamente competente One Shot™ TOP10 son (tabla 14):
| Componentes | Cantidad | Condiciones de almacenamiento |
| Células de E.coli TOP10 | 11 x 50 µl | -80ºC |
| pUC19 contol DNA (10 pg/µl): | 1 x 50 µl | -80ºC |
| Medio S.O.C | 6 ml | -80ºC |
Los componentes no suministrados son los siguientes:
– Medio LB con los antibióticos adecuados (kanamicina y cloramfenicol en nuestro caso): Comprar aquí medio LB agar con 50 µg/mL de kanamycina y 25 µg/mLcloramphenicol (nº catálogo L3230) por 82€
– Incubadora con y sin agitación a 37 °C: Comprar aquí (nº catálogo KS 4000 i control). Precio no disponible
– Baño de agua termostático: Utilizar el que se compró para la determinación de la concentración de proteínas
– Placas de petri (10 diámetro): Comprar aquí (nº catálogo P7741-1CS) por 389,00 €
– Tubos Falcon (mejores resultados que con tubos de 1,5 mL): Comprar aquí (nº catálogo 14-959-11A) por 218,53€
– Esparciador de células en forma de L: Comprar aquí (nº catálogo 14-665-231) por 69,20 €
- Réplica en placa
– Replicador para placa de Petri (Soporte + anillo) con 12 cuadrados de terciopelo estériles: Comprar aquí (nº catálogo 085884) por 319,20€
PARTE DOS: REACCIÓN LR CLONASA
- Vector de destino
El vector de destino se representa en la figura 26. Se puede comprar aquí (nº ID 1_29. Nombre del vector pK7GWIWG2(II)) por 50,00€.
Las característicasa que ofrece este vector de destino se presentan a continuación:
- Presenta dos parejas de sitios attR1 y attR2. Las parejas presentan orientación inversa entre sí. De esta manera, se recombinarán dos veces con las secuencias flanqueadas por attL1 y attL2 en el vector de entrada: una en cada una de las regiones. Esta recombinación originará la inserción de la misma secuencia en ambas regiones pero presentarán sentido opuesto, facilitando la formación de una hebra de ARN bicatenaria que es fundamento del silenciamiento mediante RNAi.
- Promotor p35S eficaz en las plantas.
- Terminador t35S, formando un ARNm estable
- Intrón en medio de las secuencias de interés, que servirá como bucle.
- Secuencia del gen ccdB en medio de las regiones originales attR1 y attR2. La expresión de este gen un inhibe la actividad de la ADN girasa, impidiendo así el crecimiento de la bacteria E.coli. Servirá como referencia de las bacterias que presenten el plásmido recombinado, ya que aquellas que no presenten el inserto, no crecerán.
- Secuencia del gen Kana, que aporta resistencia a la kanamicina en las plantas donde esté presente el plásmido. De esta manera, se podrá determinar aquellas plantas en las que se ha introducido el plásmido correctamente, ya que el tejido que no haya sido transformado, no presentará resistencia a la kanamicina y no crecerá en su presencia.
- LR clonasa
Se utilizará la mezcla de enzimas Gateway™ LR Clonase™ proporcionadas por ThermoFisher. Comprar aquí (nº catálogo 11791019) por 778,00€
La mezcla de enzimas Gateway® LR Clonase® contiene una mezcla patentada de enzimas Int (integrasa), IHF (factor de integración del huésped) y Xis (excisionasa) que cataliza la recombinación in vitro entre un clon de entrada (que contiene un gen de interés flanqueado por sitios attL, figura 27) y un vector de destino (que contiene sitios attR, figura 26) para generar su clon de expresión.
Los componentes suministrados por la mezcla de enzimas Gateway™ LR Clonase™ son:
| Componentes suminstrados | 20 rxns | 100 rxns |
| Gateway LR Clonase Enzyme Mix | 80 μl | 400 μl |
| 5X LR Clonase Reaction Buffer | 200 μl | 1000 μl |
| 2 μg/μl Proteinase K Solution | 40 μl | 200 μl |
| 50 ng/μl pENTR-gus Positive Control | 20 μl | 20 μl |
Para más información sobre los productos de la tabla 15, así como algunas recomendaciones, consultar el manual.
- Kit de purificación de plásmidos
Se utilizará el kit S.N.A.P.™ Plasmid DNA MiniPrep proporcionado por ThermoFisher. Comprar aquí (nº catálogo K190001) por 504€
Los componentes suminstrados por el kit son los siguientes:
– 100 S.N.A.P Miniprep Columns
– 100 2 ml Collection Tubes
– Resuspension Buffer (15 ml)
– RNase A, lyophilized (1.55 mg)
– Lysis Buffer (15 ml)
– Precipitation Salt (15 ml)
– Binding Buffer (60 ml)
– Wash Buffer (50 ml)
– 4X Final Wash (25 ml)
– S.N.A.P laminated manual
Los componentes no suminstrados por el kit son:
– 75 ml de etanol al 95 % para la dilución de 4X Final Wash
– Frasco o tubo esterilizado de 100 ml para dilución de 4X Final Wash
– Tubos de microcentrífuga estériles de 1,5 ml (3 por miniprep)
– Microcentrífuga
– Agua estéril o TE
PROCEDIMIENTO
Como se haintroducido anteriormente, el proceso de clonación consta de dos partes:
- INTRODUCCIÓN DEL FRAGMENTO DE ADN EN EL VECTOR DE ENTRADA
- REACCIÓN DE LA LR CLONASA
PARTE UNO
DIGESTIÓN DOBLE
Para la digestión doble del fragmento de ADN, así como del vector de entrada pENTR 2B, se seguirán los pasos descritos en el siguiente protocolo, representados en la tabla 15.
| Componentes | 50 µL de reacción |
| ADN | 1 µg |
| 10X rCutSmart Buffer | 5 µL (1X) |
| BamHI-HF | 0,5 µL (10 unidades) |
| XhoI | 0,5 µL (10 unidades) |
| Nuclease-free water | hasta 50 µL |
Ambas reacciones (digestión del fragmento de ADN y digestión del plásmido) son iguales. Por lo tanto, aunque se realizarán por separado, solo se describirán los pasos una vez.
En un tubo eppendorf de 1,5 mL:
1. Añadir 1 µg de ADN
Nota 6: La cantidad de ADN que una enzima de restricción corta depende de su aplicación. Una digestión de diagnóstico generalmente requiere ~500 ng de ADN, mientras que la clonación molecular a menudo requiere 1 µg de ADN. El volumen de reacción total suele variar entre 10 y 50 µL, según la aplicación, y está determinado en gran medida por el volumen de ADN que se va a cortar. Por otro lado, la cantidad de ADN también depende de la intensidad de la banda cuando se hizo el gel de agarosa (PASO 4. PURIFICACIÓN DEL FRAGMENTO DE ADN).
Para añadir 1 µg del fragmento de ADN se debe tener en cuenta:
– Nanodrop mide 2 nanogramos (0,002 µg)
– El protocolo de PCR se realiza en un volumen de 25 µl (tabla 9)
0,002 µg –> 25 µl
1 µg –> x µl
x = 0,05 µl = 50 nL
Para añadir 1 µg del vector de entrada se debe tener en cuenta:
– ThermoFisher proporciona 10 µg de plásmido
– El vector llega en 20 µl en tampón TE, pH 8,0.
10 µg –> 20 µl
1 µg –> x
x = 2 µl
2. Añadir 5 µl de 10x rCutSmart buffer
Nota 7: Se recomienda un volumen de reacción de 50 µl para la digestión de 1 µg de sustrato. Si se pretende cambiar el volumen total, el volumen del buffer, o la cantidad de ADN, consultar la tabla 16. Para más información, consultar el siguiente enlace

3. Añadir 0,5 µl de cada enzima de restricción
Nota 8: En el protocolo original se especifica 1,0 µl, pero en este caso, este volúmen ha sido ajustado. En general, se recomienda de 5 a 10 unidades de enzima por µg de ADN y de 10 a 20 unidades de ADN genómico en una digestión de 1 hora. El volumen de la enzima no debe exceder el 10 % del volumen total de la reacción para evitar la actividad estelar debida al exceso de glicerol.
4. Añadir 44 µl de nuclease-free water
5. Incubar a 37 ºC durante 5-15 minutos
Nota 9: Las enzimas de restricción utilizadas son «Time-saver qualified». Para más información consultar el siguiente enlace
6. Purificación del ADN (electroforesis)
PURIFICACIÓN DEL ADN
Se seguirá el mismo protocolo que en el PASO 4. PURIFICACIÓN DEL ADN.
Después de la doble digestión del vector y del inserto, es importante someter a los fragmentos deseados a un proceso de purificación, a través de un gel de agarosa. El gel de agarosa, además, elimina los restos de enzimas y sales presentes tras la reacción de digestión enzimática.
Se podrían recuperar los ácidos nucleicos del gel de agarosa por el método tradicional, que consiste en la extracción de los fragmentos por fenol/cloroformo y la posterior preciitación por etanol. Sin embargo, este método reduce demasiado el rendimiento de la purificación.
Debido a que la reacción de ligación posterior requiere fragmentos de ADN puros, se medirá el ratio A260/A230 > 2.0 y el ratio A260/A280 > 1.8, con el objetivo de determinar la pureza de los fragmentos de ADN. Para ello, se utilizará el intrumento Nanodrop (PASO 2. DISEÑO DE CEBADORES) y se seguirán los pasos descritos en el vídeo 3.
REACCIÓN DE LIGACIÓN
Para la reacción de ligación se seguirán los pasos descritos en el siguiente protocolo, representado en la tabla 17.
| Componentes | 20 µl de reacción |
| Tampón de ADN ligasa T4 (10X) | 2 μl |
| vector de ADN (3,8 kb) | 50 ng |
| Inserto de ADN (288 pb) | 11,05 ng |
| Nuclease-free water | to 20 μl |
| T4 DNA Ligasa | 1 μl |
La reacción descrita se realizará en un tubo eppendorf de 1,5 mL con hielo. La T4 DNA ligasa se añade al final. Por otro lado, el T4 DNA Ligase Buffer debe descongelarse y resuspenderse a temperatura ambiente.
- Mezcle suavemente la reacción pipeteando hacia arriba y hacia abajo y microcentrífuga brevemente.
- Para extremos romos o salientes de una sola base, incube a 16 °C durante la noche o a temperatura ambiente durante 2 horas (alternativamente, se puede usar una ligasa de ADN T4 de alta concentración en una ligadura de 10 minutos).
- Inactivación por calor a 65°C durante 10 minutos. Enfríe en hielo y transforme 1-5 μl de la reacción en 50 μl de células competentes
TRANSFORMACIÓN DE BACTERIAS
La transformación es un proceso natural en el que las células bacterianas captan ADN extraño a baja frecuencia. En biología molecular, este proceso se utiliza para propagar plásmidos dentro de bacterias que se han vuelto «competentes» (porosas) para la absorción de ADN. Las células competentes están comercialmente disponibles en el siguiente enlace, y se describirán más adelante.
Una forma de tratar las bacterias para que sean competentes para la transformación es someterlas a un choque térmico: Para ello, las bacterias en fase logarítmica se tratan con cloruro de calcio. Posteriormente, se mezclan con el ADN resultante de la reacción de ligación y se someten a un choque térmico de 42 ºC. Parte del ADN será absorbido por las bacterias, y comenzará a replicarse. Otra forma de transformación es la electroporación, pero no será el tratamiento que se utilice en este trabajo. En el vídeo 6 se describen algunos conceptos básicos para la transformación de células competentes que se deben tener en cuenta.
Las bacterias transformadas (después del choque térmico o la electroporación) se colocan en una placa de agar con un antibiótico apropiado y se analizan mediante el método adecuado que permita distinguir colonias que lleven el plásmido deseado con el inserto.
El cribado de bacterias para la identificación de las bacterias transformadas se puede hacer de diferentes maneras, tales como incubación con antibióticos, selección positiva (crecen las que no puedan expresar un gen letal), digestión con enzimas de restricción, PCR, secuenciación de Sanger, etc. En este trabajo sólo se realizará la selección de bacterias transformadas a través de la incubación con antibióticos y la selección positiva, debido a que el plásmido de entrada utilizado (figura 27) lo permite. Para más inforamción sobre otrass técnicas de análisis consultar el siguiente enlace.
Una vez introducida la transformación de células competentes se procede a desribir cada paso clave:
- Preparación de células competentes
E.coli es la especie bacteriana conmunmente utilizada, pero dado que la competencia natural de E. coli es muy baja o incluso inexistente, las células deben volverse competentes para la transformación por choque térmico.
Las células competentes prefabricadas están disponibles en formatos listos para usar de fuentes comerciales. Estas células competentes tienen control de calidad y se prueban para cumplir con las especificaciones de eficiencia de transformación y genotipos . Estas preparaciones minimizan la variabilidad de lote a lote y simplifican significativamente la propagación eficiente del ADN clonado.
Se utilizarán las células competentes descritas en el apartado de MATERIALES de esta misma sección.
- Transformación
1. Antes de comenzar
- Equilibrar un baño de agua a 42°C
- Calentar el vial del medio S.O.C a temperatura ambiente
- Caliente las placas selectivas en una incubadora a 37 °C durante 30 minutos (utilice una placa para cada transformación). Se utilizarán 11 placas.
- Preparar las placas del medio LB con antibiótico
2. Procedimiento para la transformación química (choque térmico)
Primero, las células se incuban con ADN en hielo durante 5 a 30 minutos en un tubo de polipropileno. Deben evitarse los tubos de poliestireno, ya que el ADN puede adherirse a la superficie, lo que reduce la eficiencia de transformación.
- Centrifugue los viales que contienen la reacción de ligación e incúbelos en hielo (11 viales)
- En hielo se incuban cada uno de los viales One Shot® de células (50 µl) (11 viales)
- Pipetea 1-5 µl de cada reacción de ligación en cada vial de células competentes y agite. No mezclar pipeteando hacia arriba y hacia abajo. La mezcla de ligación que sobre se puede almacenar a -20ºC
- Incubar los viales en hielo durante 30 minutos.
- Incubar 30 segundos a 42 ºC en el baño térmico
- Introducir los viales en hielo
- Período de recuperación celular
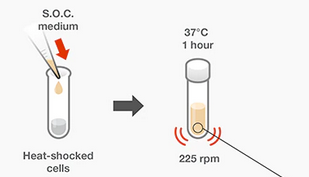
Después del choque térmico o la electroporación, las células transformadas se cultivan en un medio líquido libre de antibióticos durante un período corto para permitir que comience la expresión de los genes de resistencia a los antibióticos del plásmido adquirido (figura 28). Se recomienda el medio SOC, que contiene glucosa y MgCl2, para maximizar la eficiencia de transformación.
- Añadir 250 µl del medio S.O.C pre-calentado a los viales que estaban en hielo.
- Coloque los viales en una gradilla de microcentrífuga de costado y asegúrelos con cinta adhesiva para evitar que se pierdan. Agite los viales a 37 °C durante exactamente 1 hora a 225 rpm en una incubadora con agitación.
 |
 |
- Recubrimiento celular
Después de crecer en medio SOC, las células se sembraron en agar LB con los antibióticos apropiados u otros agentes para la identificación y recuperación de transformantes exitosos. Antes de sembrar las células, las placas se deben precalentar a una temperatura de crecimiento favorable y deben estar libres de condensación para evitar la contaminación y la mezcla de colonias.
La cantidad de células sembradas en placas debe producir un número suficiente (y tampoco demasiado numeroso) de colonias distintas e individuales para la detección posterior. Las células cultivadas en medio SOC pueden sedimentarse mediante centrifugación durante 5 minutos a 600–800 x g y resuspenderse en un volumen más pequeño para sembrar en placas. Para sembrar en una placa de 100 mm, 100–200 μL de suspensión celular generalmente funcionan bien.
- Extienda de 20 a 200 μL de cada vial de transformación en placas de agar LB separadas y etiquetadas. La mezcla de transformación restante puede almacenarse a 4 °C y colocarse en placas al día siguiente, si se desea.
- Invierta las placas e incube a 37°C durante la noche.
- Seleccione colonias y analice mediante aislamiento de plásmidos, PCR o secuenciación.
- Análisis y selección de colonias
Las bacterias sin el vector carecen del gen de resistencia a los antibióticos y no crecerán, mientras que las bacterias transformadas con el vector sobreviven debido al gen de resistencia a los antibióticos expresado (figura 29).

En el caso de nuestro trabajo, el vector sin inserto no crecerá (a diferencia del esquema de la figura 29), debido a la presencia del gen ccdB en el vector. Cuando no esté el inserto pero sí esté presente el vector (figura 27), se expresará el gen ccdB, impidiendo el crecimiento celular.
De esta manera, y de acuerdo con la figura 29, podemos obtener las siguientes situaciones:
- Células sin vector (no observaremos crecimiento, por falta de resistencia a antibióticos)
- Células con vector con reacción de ligación errónea (no observaremos crecimiento, por falta de resistencia a antibióticos)
- Células con inserto sin vector, por ligación errónea (no observaremos crecimiento, por falta de resistencia a antibióticos)
- Células con vector sin inserto (no observaremos crecimiento, por la expresión del gen ccdB presente en el vector)
- Células con vector y con inserto indeseado (observaremos crecimiento, si el inserto se encuentra en la región flanqueada por attL1 y attL2)
- Células con vector y con inserto deseado (observaremos crecimiento)
1. PCR para la identificación de cepas bacterianas transformadas
En este senitdo, debe hacerse un análisis un poco más profundo (no basta con el cribado por resistencia a antibióticos o selección positiva, debido a que puede darse la situación número 5. Se realizará una PCR siguiendo las mismas etapas y materiales que en el PASO 3. AMPLIFICACIÓN DEL ADN.
En la figura 30 encontramos los posibles enfoques para el diseño de cebadores en la detección mediante PCR para la identificación de cepas bacterianas con el inserto de interés. En este caso, se utilizarán los mismos cebadores que en la amplificación del ADN del paso 3 (cebadores específicos del inserto)

En esta reacción de PCR no se tendrá en cuenta Tm del vector, debido a que el objetivo de esta reacción no lo necesita. Con esta PCR sólo se pretende detectar la presencia o ausencia de una banda en el gel de agarosa, como resultado de la presencia o ausencia del inserto en el vector.
La Tm influye, sobretodo, en el rendimiento y especificidad de la reacción de PCR, para otros objetivos tales como la cuantificación de ADN, la determinación de la expresión de un gen, la detección de mutaciones, etc.
2. Réplica en placa
Importante: Este paso debe realizarse antes de efectuar la PCR descrita anteriormente.
La réplica en placa es una técnica comúnmente utilizada en biología molecular y microbiología, y consiste en la transferencia del contenido presente en la placa maestra a una (o varias) segunda placa, con el objetivo de poder comparar las colonias, o someterlas a una reacción de PCR, sin perder la colonia de partida (placa maestra). Para más información sobre esta técnica visitar el siguiente enlace.
El procedimiento que se seguirá será el descrito en el vídeo 7
3. Electroforesis en gel de agarosa
Tras la PCR se realizará una electroforesis en gel de agarosa, siguiendo los mismos pasos que los descritos en el PASO 4. PURIFICACIÓN DEL ADN, con el objetivo de determinar la presencia o ausencia del inserto, al comparar las bandas obtenidas tras la reacción de PCR con un control negativo (bacterias a las que no se introdujo ningún plásmido).
De esta froma, deberá aparecer una banda si está presente el inserto (habrá sido amplificado el inserto gracias a los cebadores específicos, figura 30), de ~280-250 pb. Si no está presente el inserto deseado, no debe aparecer ninguna banda en el gel.
PARTE DOS
PURIFICACIÓN DEL PLÁSMIDO DEL CLON DE ENTRADA
Antes de comenzar con la reacción de la LR clonasa, debemos purificar el vector de entrada de las bacterias que mostraron la presencia del inserto en el vector, tras la electroforesis realizada anteriormente. Para ello, se seguirá el protocolo siguiente:
- Antes de comenzar
- Resuspender todo el contenido de ARNasa A (1,55 mg) en 200 μl de tampón de resuspensión y añádalo al tampón de resuspensión restante. El tampón de resuspensión debe almacenarse ahora a +4ºC.
- Agregue los 25 ml de 4X Final Wash a 75 ml de etanol al 95% para hacer 100 ml de Lavado Final 1X
- Compruebe el tampón de lisis para ver si hay un precipitado blanco. Si está presente, coloque el tampón en un baño de agua a 37ºC durante 5 minutos hasta que la solución se aclare.
- Lisis y precipitación
- Centrifugar 1-3 ml de un cultivo durante la noche para sedimentar las células. Usar celda
cultivos que son de 1 a 1,5 x 109 células/ml. - Vuelva a suspender el sedimento celular en 150 μl de tampón de resuspensión agitando en vórtex o pipeteando suavemente hacia arriba y hacia abajo.
- Añadir 150 μl de Lysis Buffer y mezclar suavemente invirtiendo el tubo 5-6 veces. Incubar durante 3 minutos a temperatura ambiente
- Añadir 150 μl de sal de precipitación helada e invertir 6-8 veces para asegure una mezcla completa de todos los componentes.
- Centrifugar en una microcentrífuga a temperatura ambiente a 14.000 x g durante 5 minutos.
- Mientras espera la centrífuga, coloque el S.N.A.P. Columna Miniprep (A) dentro del tubo de recolección de 2 ml provisto (B), como se muestra en la figura 31.

- Unión de plásmidos
- Pipetea el sobrenadante en un tubo de microcentrifuga estéril. Descarta el pellet gelatinoso
- Agregue 600 μl de Binding Buffer y mezcle invirtiendo 5-6 veces. Pipetee o vierta toda la solución en el tubo de recogida S.N.A.P. Miniprep
- Centrifugue la columna/tubo de recogida S.N.A.P.™ Miniprep a temperatura ambiente a 1000-3000 x g durante 30 segundos. Alternativamente, deje que el tampón de unión fluya a través de la columna por gravedad durante 10-15 minutos. El ADN del plásmido ahora está unido a la columna
- Deseche el flujo de la columna.
- Añada 500 μl de Wash buffer
- Centrifugue la columna/tubo de recolección S.N.A.P.™ Miniprep a temperatura ambiente a 1.000-3.000 x g durante 10-30 segundos o dejar escurrir por gravedad durante 7 minutos. Deseche el flujo de la columna.
- Agregue 900 μl de 1X Final Wash y centrifugue (el lavado final 1X se realizó en Antes de comenzar, Paso 2).
- Centrifugue la columna/tubo de recolección S.N.A.P.™ Miniprep a temperatura ambiente a máxima velocidad durante 1 minuto para secar la resina.
- Elución del plásmido
- Para eluir el ADN plasmídico, transfiera la columna S.N.A.P.™ Miniprep a un tubo de microcentrífuga estéril y agregue 60 μl de tampón TE o agua estéril directamente a la resina. Incubar durante 3 minutos a temperatura ambiente.
- Centrifugue la columna/tubo de recolección S.N.A.P.™ Miniprep a temperatura ambiente a máxima velocidad durante 30 segundos. El ADN del plásmido es ahora eluido de la columna. Retire y deseche la columna.
- Determinación de la pureza y concentración
Para determinar la concentración y pureza del plásmido se utilizará Nanodrop (vídeo 3)
REACCIÓN LR CLONASA
Una vez que haya obtenido un clon de entrada que contenga su gen de interés, puede realizar una reacción de recombinación LR entre el clon de entrada y un vector de destino, y transformar la mezcla de reacción en un huésped E. coli adecuado (las mismas células competentes que se utilizaron en la PARTE UNO)
Utilice el siguiente procedimiento para realizar una reacción de recombinación LR. Para
control positivo, use 100 ng (2 μl) de pENTR-gus.
- Agregue los componentes de la tabla 18 a un tubo de microcentrífuga de 1,5 ml a temperatura ambiente y mezcla
- Retire la mezcla de enzimas LR Clonase de -80 °C y descongélela en hielo durante unos 2 minutos. Agite en un vórtex la mezcla de enzimas clonasa LR brevemente dos veces (2 segundos cada vez).
- A cada muestra, agregue 4 μl de mezcla de enzimas LR Clonase a la reacción y mezcle bien agitando brevemente dos veces. Microcentrífuga brevemente.
- Regrese la mezcla de enzimas LR Clonase a -80°C inmediatamente después de su uso.
- Incuba las reacciones a 25ºC durante 60 minutos
- Agregue 2 μl de la solución de proteinasa K a cada muestra para terminar elreacción. Agite en el vórtex brevemente. Incube las muestras a 37 °C durante 10 minutos.
TRANSFORMACIÓN DE BACTERIAS
La transformación de bacterias se realizará siguiendo los mismos pasos que en la PARTE UNO, descrita anteriormente.
En el análisis y selección de colonias debemos tener en cuenta que sólo crecerán aquellas bacterias que hayan introducido el inserto en el plásmido en dos sitios: un sitio flanqueado por attR1 y attR2 y otro sitio flanqueado por attR1 y attR2 en sentido opuesto. Si alguno de estos sitios no ha sido sustituido por el inserto, la expresión del gen ccdB no permitirá el crecimiento de las células.
Nota 10. Para hacer posible la selección con el gen ccdB se han escogido cepas de E.coli que no contienen el episoma F’ (TOP10). Sin embargo, algunas cepas de E.coli parecidas, pueden contener el episoma F’ (TOP10 F’), por lo que habrá que tener precaución a la hora de escoger las cepas. Aquellas que contienen el episoma F’ contienen el gen ccdA
PASO 6. TRANSFORMACIÓN DE LAS PLANTAS DE AGUACATE
INTRODUCCIÓN
Para utilizar los mecanismos de mejoramiento genético in vitro se requiere tener previamente el sistema de regeneración de una planta completa a través de embriogénesis somática u organogénesis adventicia. La regeneración del aguacate por embriogénesis somática se ha descrito en diferentes variedades como Hass, Duke, T372, Anaheim, Duke 7 y Reed. Para estudiar la vía de regeneración embriogénica del aguacate se han utilizado diferentes explantes, como embriones zigóticos inmaduros en estadio globular o nucela.
Por otro lado, la transformación transitoria del callo embriogénico del aguacate se realizó a través de ADN biolístico, un método de transferencia directa de genes en una célula, creando así organismos transgénicos, comunmente utilizado para manipular organismos vegetales. Consiste en propulsar los genes de interés dentro de la célula con la ayuda de un cañon de ADN, con lo cual se modifica el ADN de las células vegetales. Antes del uso, los parámetros físicos y biológicos para el bombardeo de partículas deben optimizarse para cada tejido en particular.
MATERIALES
PURIFICACIÓN DEL PLÁSMIDO DE DESTINO
El mismo kit de purificación de plásmidos que se utilizó en la reaccion de LR clonasa (Ver PASO 5. SISTEMA DE CLONACIÓN)
REGENERACIÓN DE PLANTAS DE AGUACATE
– Cultivos embriogénicos de aguacate, cv. ‘Duke 7’, se establecieron a partir de embriones cigóticos inmaduros (ver protocolo de regeneraciín de plantas)
– Medio de cultivo Murashige y Skoog (MS). Comprar aquí (nº catálogo 10494). Precio no disponible
– Picloram. Comprar aquí (nº catálogo 36774-250MG-R) por 78,70 €
– Agar (6 g/l). Comprar aquí (nº catálogo A1296-100G) por 78,30 €
ADN BIOLÍSTICO
- Bombardeo de partículas
– PDS-1000/He™ System and Hepta™ Systems. Comprar aquí. Precio no disponible
– Helios® Gene Gun System. Comprar aquí. Precio no disponible

– Cilindro de gas helio; alta presión (2400–2600 psi [16 547,4–17 926,4 kPa]); grado 4.5 o 5.0 (99.995% o mayor pureza). Comprar aquí (nº catálogo HE HP300). Precio no disponible
– Caja de plástico opaco esterilizada con etanol al 70% para almacenar placas bombardeadas.
– Tamices de parada (Bio-Rad), esterilizar en autoclave. Comprar aquí (nº catálogo 1652336) por 309,00€
– Bomba aspiradora; paleta rotativa llena de aceite, con una velocidad de bombeo de 90–150 L/min (3–
5 pies 3 /min)
- Preparación de microportadores recubiertos de ADN
Para la esterilización de macroportadores:
– Macroportadores para dispositivo biolístico (Bio-Rad).
– Porta macroportadores (Bio-Rad).
– 70 y 95% etanol.
– Vaso de precipitados de vidrio y placa Petri de vidrio (tratados en autoclave).
– toallas de papel
– Fórceps estériles con puntas finas (las puntas curvas funcionan bien).
– Desecante en placas Petri de vidrio. Un papel de filtro estéril o una placa de Petri de plástico invertida
con agujeros (hecho en casa) debe colocarse sobre el desecante para proporcionar una plataforma libre de polvo para cargar partículas recubiertas de ADN en los macroportadores. En el protocolo se usa el desecante de la marca Drierite, que cambia de azul a rosa a medida que absorbe agua. Se debe hornear a 180 °C durante aproximadamente 4 h para restaurar el color azul y la capacidad de desecación.
Para la esterilización de partículas de oro:
– Microportadores: partículas de oro de 0,75 μm de diámetro (Analytical Scientific Instrument, El Sobrado, CA)
– Pequeño vial o tubo de vidrio (1–3 mL).
– Horno que alcanzará los 180°C.
– Isopropanol, grado HPLC.
– Glicerol (50% v/v): Mezclar glicerol 1:1 con agua tipo I y autoclave.
Para el recubrimiento de partículas de oro con ADN
– Micropipetas y puntas (rango de 5 a 500 μL).
– ADN de plásmido (1 μg/μL) en tampón TE estéril (Tris-HCl 1 mM, pH 7,8, 0,1 mM
ácido etilendiaminotetraacético disódico [EDTA])
– CaCl2 2,5 M, esterilizado por filtración: Para hacer 50 mL, disolver 18,38 g de cloruro de calcio dihidrato en agua tipo I. Esterilizar por filtración y almacenar a 4°C en pequeñas alícuotas
– Spermidina (Sigma-Aldrich nº catálogo S0266). Agregue 1 ml de agua tipo I, ajuste el volúmen a 68,9 ml, agite y esterilice por filtración. Almacenar a -20ºC en alícuotas de 1,2 ml (puede almacenarse durante un mes)
– Isopropanol grado HPLC
– Limpiador ultrasónico de baño de agua (Modelo B1200R-1; Branson Ultrasonics Corporation)
tion, Danbury, CT, o unidad similar).
SELECCIÓN Y GERMINACIÓN
– Solución madre de monosulfato de kanamicina (Km) (25 mg/ml, pH 5,8, esterilizado por filtración). Prepare en agua tipo I. Conservar a –20°C en pequeñas alícuotas. El caldo congelado es bueno indefinidamente. Caliente para agregar a los medios esterilizados en autoclave que tiene enfriado a 50-55°C.
– Medio selectivo: medio MS-HF 1/2 con 30 g/L de sacarosa, 3 g/L de activado carbón, 7 g/L de Bacto-agar (Difco, Detroit, MI) y 10 o 15 mg/L de Km (agregado después del autoclave). Ajuste el pH a 5,8 con KOH y autoclave. Dispensar en alícuotas de 20 ml en placas Petri de 100 × 15 mm.
PROCEDIMIENTO
PURIFICACIÓN DEL PLÁSMIDO DE DESTINO
Se seguirá el protocolo proporcionado por el kit, descrito en la en la reaccion de LR clonasa (Ver PASO 5. SISTEMA DE CLONACIÓN) y detallado en el siguiente enlace.
REGENERACIÓN DE PLANTAS DE AGUACATE
Se seguirá el protocolo detallado en el siguiente enlace.
ADN BIOLÍSTICO
El procedimiento se basa en las explicaciones del vídeo 8, donde se decribe el sistema de transfección de partículas biolísticas Helios Gene Gun. Además, incluye la configuración del instrumento, la preparación del microportador y el cartucho, y el funcionamiento del instrumento.
Para este proceso se seguirá el protocolo detallado en el siguiente enlace. Sin embargo, también habrá que tener en cuenta la optimización del proceso para una planta de aguacate, siguiendo el siguiente enlace.
SELECCIÓN Y GERMINACIÓN
Se seguirá el protocolo detallado en el siguiente enlace.
PASO 7. CARACTERIZACIÓN DE LA ENZIMA. ACTIVIDAD ENZIMÁTICA DE PPO EN AGUACATE y rt-pcr
INTRODUCCIÓN
Con el objetivo de comprobar el efecto del iRNA:
1. Se medirá la actividad enzimática de la PPO en plantas de aguacate antes y después de la inoculación de siRNA. Para ello, se seguirá el protocolo que acompaña al kit con nº de referencia E-BC-K259-M-96T. El precio de este kit no está disponible en la página de QuimiGen, pero se puede consultar en el siguiente correo electrónico <pedidos@quimigen.com>.
A continuación, se decribe un protocolo para medir la actividad enzimática de la PPO.
PRINCIPIO DE DETECCIÓN
Este kit está basado en un método colorimétrico y contiene como instrumento de detección un lector de microplacas (405 nm-415 nm, longitud de onda óptima: 410 nm). Se pueden detectar 48 muestras sin duplicación.
La PPO puede catalizar compuestos fenólicos en sustancias de quinona. Este último tiene absorción específica a 410 nm. Por lo tanto, la actividad de la PPO se puede calcular indirectamente midiendo el valor OD a 410 nm.
MATERIALES
A continuación, se presentan los materiales proporcionados y no proporcionados por el kit nº E-BC-K259-M-96T que se pretende utilizar (figura 33)

- Componentes proporcionados por el kit
Se muestran en la tabla 18.
| Ítem | Componentes | Especificación | Almacenamiento |
| Reactivo 1 | Solución de extracción | 60 ml x 2 viales | 2-8 ºC, 6 meses |
| Reactivo 2 | Buffer | 40 ml x 2 viales | 2-8 ºC, 6 meses |
| Reactivo 3 | Sustrato | 20 ml x 1 vial | 2-8 ºC, 6 meses , luz de sombreado |
| Microplato | 96 pocillos | ||
| sellador de placas | 2 piezas |
- Componentes no proporcionados por el kit
– Lector de microplacas (410 nm): Comprar aquí S6 Ultimate M2. Precio no disponible
– Tubos eppendorf (1,5 ml): Comprar aquí (nº catálogo 3451PK) por 19,40€
– Tubos eppendorf (2 ml): Comprar aquí (nº catálogo 3453PK) por 23,00€
– Mezclador vórtex: Comprar aquí (nº de catálogo 88882010) por 388€
– Baño de agua termostático: Comprar aquí. Precio no disponible
– Puntas de micropipeta (100-1000 μL): Comprar aquí (nº catálogo CLS4868-1000EA) por 38,70€
– Puntas de micropipeta (0’5-10 μL) Comprar aquí (nº catálogo CLS4830-960EA) por 101,00€
– Agua destilada (1L): Comprar aquí (nº catálogo 8483331000) por 25,90€
– Micropipetas (100-1000 μL): Comprar aquí (nº catálogo 10030259) por 69,00€
– Micropipetas (0’5-10 μL): Comprar aquí (nº catálogo 17014388). Precio no disponible
PROCEDIMIENTO
A continuación, se describe el protocolo para el análisis de la actividad enzimática de PPO proporcionado por el kit nº E-BC-K259-M-96T. Para más información, visitar la página de QuimiGen
PUNTOS CLAVE DEL ENSAYO
- La temperatura de incubación debe ser de 37 ºC
- Se recomiendan los tubos eppendorf a prueba de explosiones para el baño en agua caliente (100 ºC).
- La aparaición de sustancias en suspensión en algunos tubos es un fenómeno normal. Se puede centrifugar y coger el sobrenadante para medir el valor OD
PREPARACIÓN DE REACTIVOS
- Precaliente el reactivo 1 (solución de extracción) a 37ºC por 20 minutos. Úsalo después de aclarar completamente.
- Mantener el reactivo 2 y 3 (buffer y sustrato, respectivamente) a temperatura ambiente.
PREPARACIÓN DE LA MUESTRA
1. Solución A. Extracción del extracto crudo de enzima
- Pese con precisión la muestra de tejido de la planta
- Agregue el reactivo 1 de acuerdo con la proporción de Peso (g): Volumen (mL) =1 :9.
- Homogeneizar mecánicamente la muestra en baño de agua con hielo.
- Centrifugue a 11000g durante 15 min.
- Lave el sobrenadante para la detección.
- Mientras tanto, determine la concentración de proteínas del sobrenadante (E-BC-K318-M).
2. Solución B. Extracción del extracto crudo de enzima (tubos control)
- Después de extraer la solución enzimática cruda A, se lavó el 50% del sobrenadante en un tubo EP nuevo de 1,5 mL.
- Se hirvió a 100ºC durante 5 min.
- Enfriar los tubos con agua corriente
DILUCIÓN DE LA MUESTRA
Se recomienda tomar 2-3 muestras con una gran diferencia esperada para hacer un pre-experimento antes del experimento formal y diluir la muestra de acuerdo con el resultado del pre-experimento.

CONFIGURACIÓN DE LA PLACA
En la figura 34 se observa la configuración de la placa para el ensayo de la actividad enzimática, donde S1-S48 son los pocillos de muestra, y S1′-S48′ son los pocillos de muestras control.

OPERACIONES
- Tubo control: Añadir 600 μL de reactivo 2 en tubos eppendorf de 1,5 ml.
- Tubo de muestra: Añadir 600 μL de reactivo 2 en tubos eppendorf de 1,5 ml.
- Añadir 150 μL del reactivo 3 en cada uno de los dos tubos eppendorf anteriores.
- Tubo control: Añadir 150 μL de extracto crudo de enzima (solución B).
- Tubo de muestra: Añadir 150 μL de extracto crudo de enzima (solución A).
- Mezclar con el vórtex, incubar a 37 ºC durante 3 minutos. Inmediatamente después, incubar a 100 ºC en el baño de agua caliente durante 5 minutos. Después, enfriar los tubos a temperatura ambiente.
- Introducir 320 μL de los tubos en el microplato y medir el valor OD para cada pocillo a 410 nm. De esta manera, obtendremos un valor de OD de la muestra (A1) y un valor de OD del tubo control (A2). Luego, el incremento de absorbancia será la diferencia entre ambos valores (A1-A2).
CÁLCULO DE LA ACTIVIDAD ENZIMÁTICA
El cálculo de la actividad enzimática se realiza siguiendo la ecuación 2, teniendo en cuenta los siguientes datos:
– ΔA = A1 – A2
– V: Volumen de la muestra añadida a la reacción (0,15 mL)
– T: tiempo de reacción (3 min)
– Cpr: Concentración de proteína en la muestra (para conocer este valor debemos realizar el protocolo descrito a continuación).
– f: factor de dilución de la muestra antes de ser evaluada

DETERMINACIÓN DE LA CONCENTRACIÓN DE PROTEÍNA
A continuación, se describe el protocolo para la determinación de la concentración de proteínas, necesario para el cálculo de la actividad enzimática. Se seguirá el protocolo proporcionado por el kit nº E-BC-K318-M, propuesto por el mismo protocolo anterior. El precio de este kit es de 141,34€
PRINCIPIO DE DETECCIÓN
Este kit está basado en un método colorimétrico y contiene como instrumento de detección un lector de microplacas. Se pueden detectar 80 muestras sin duplicación. Además, presenta una sensibilidad de 0.0165 mg/mL y un rango de detección de 0.0165-1 mg/mL.
Este kit se puede utilizar para medir el contenido de proteína total (TP) en suero, plasma, cultivo de células, tejido y muestras celulares. El kit llamado «concentración de proteína BCA» es un método para la determinación de la concentración de proteínas rápido, estable, sensible y válido para cualquier tipo de proteína. Además, no se ve afectada por las sustancias químicas de la muestra, que pudieran interferir.
El principio de detección se basa en que el Cu2+ puede ser reducido a Cu+ por la proteína, en un medio alcalino. Cu+ se puede combinar con el reactivo BCA y formar un complejo púrpura, que tiene un pico de absorción máximo a 562 nm. El valor de absorbancia es proporcional a la concentración de proteína.
Por lo tanto, la concentración de proteína se puede calcular de acuerdo con el valor de OD (figura 35)

MATERIALES
- Componentes proporcionados por el kit
Los componentes proporcionados por el kit se muestran en la tabla 20.
| Ítem | Componente | Especificación | Almacenamiento |
| Reactivo 1 | Reactivo BCA | 25 mL × 1 vial | 12 meses |
| Reactivo 2 | Solución de sal de cobre | 0.5 mL × 1 vial | 12 meses |
| Reactivo 3 | Estándar de proteína BSA | 1 mg × 1 vial | 12 meses |
| Reactivo 4 | Diluyente estándar | 15 mL × 1 vial | 12 meses |
| Microplato | 96 pocillos | No se requiere | |
| sellador de placas | 2 piezas |
- Componentes no proporcionados por el kit
Los componentes no proporcionados por el kit son los siguientes:
– Lector de microplacas (540-590 nm): Utilizar el que se compró para el protocolo de la actividad enzimática
– Mezclador vórtex: Utilizar el que se compró para el protocolo de la actividad enzimática
– Micropipetas (10, 200, 1000 μL): Utilizar las que se compraron para el protocolo de la actividad enzimática
– Incubadora de laboratorio: Comprar aquí (nº catálogo EX10030737) por 269,00 €
– Puntas de micropipeta (10 μL, 200 μL, 1000 μL): Utilizar las que se compraron para el protocolo de la actividad enzimática
– Tubos eppendorf (1.5 mL, 2 mL, 5 mL): Utilizar los que se compraron para el protocolo de la actividad enzimática
– ddH2O: Utilizar la que se compró para el protocolo de la actividad enzimática
– Solución de NaCl (1L, 0,9%): Comprar aquí (nº catálogo 567442-1EA) por 38,70 €
– PBS (0,01 M, pH 7,4): Comprar aquí (nº catálogo P3813-1PAK) por 14,60 €
PROCEDIMIENTO
PUNTOS CLAVE DEL ENSAYO
- La concentración de la muestra de proteína debe diluirse a 1 mg/mL o menos, con la solución salina normal (0,9% NaCl). Mostrará un buen rango lineal por debajo de este concentración.
- Evitar la formación de burbujas al añadir los reactivos al microplato
PREPARACIÓN DE REACTIVOS
1. Preparación de soluciones de trabajo BCA
- Mezcle completamente el reactivo 1 y el reactivo 2 en una proporción de 50:1.
- Prepara la cantidad necesaria de solución antes de su uso.
- La solución de trabajo preparada se puede almacenar a 4°C durante 24 h.
2. Preparación de solución estándar de 1 mg/mL
- Disuelva un vial de reactivo 3 en polvo con 1 mL de reactivo 4.
- Mezcle completamente antes de usar.
- Se recomienda dividir en alícuotas la solución preparada. Se puede almacenar a -20°C durante 3 meses
PREPARACIÓN DE LA MUESTRA
En este caso, se tienen muestras de tejidos que serán preparadas como se explica a continuación. En el caso de que la muestra fuera de otro tipo, el método a seguir sería diferente. Para más información consultar el protocolo proporcionado por el kit.
- Tome 0,02-1 g de tejido fresco para lavar con el medio de homogeneización a 2-8 °C.
- Absorber el agua con papel filtro y pesar.
- Homogeneizar en la proporción del volumen de medio homogeneizado (2-8°C ) (mL): el peso del tejido (g) =9:1.
- Luego, centrifugar el homogeneizado de tejido durante 10 min a 10000 g a 4 °C.
- Toma el sobrenadante para conservarlo en hielo para su detección.
- Si no se detecta el mismo día, la muestra de tejido (sin homogeneización) se puede almacenar a -80°C durante un mes.
Nota 11: El medio de homogeneización es PBS (0.01 M, pH 7) o 0,9% NaCl. El método de homogeneización es el siguiente: (1) Manual: Pesar el tejido y picarlo en trozos pequeños (1 mm3), luego coloque las piezas de tejido en un tubo homogeneizado de vidrio. Añadir el medio homogeneizado en un tubo homogeneizado. Coloque el tubo en el baño de hielo con la mano izquierda, e inserte la varilla de apisonamiento de vidrio verticalmente en el tubo homogeneizado con la mano derecha para moler hacia arriba y hacia abajo durante 6-8 min. O coloque el tejido en el mortero y agregue nitrógeno líquido para moler completamente. Entonces, añadir el medio homogeneizado para homogeneizar. (2) Homogeneizado mecánico: Pesar el tejido en el tubo EP, añadir el medio homogeneizado para homogeneizar el tejido con instrumento homogeneizador (60 Hz, 90 s) en el baño de hielo. (Para muestras de piel, músculo y tejido vegetal, el tiempo de homogeneización se puede prolongar adecuadamente). (3) Ultrasonicación: tratar las células con disruptor celular ultrasónico (200 W, 2 s/tiempo, intervalo de 3 s, en un tiempo total de 5 min)
- Consideraciones en la preparación de muestra
- Si se utiliza un tampón de lisis para preparar homogeneizados de tejidos o cultivos celulares
sobrenadante, existe la posibilidad de causar una desviación debido a la sustancia química introducida. - Se debe predecir la concentración antes del ensayo. Si la concentración de la muestra no está dentro del rango de la curva estándar, los usuarios deben determinar las diluciones de muestra óptimas para su experimento particular.
- La muestra no debe contener agentes quelantes (EGTA, EDTA) ni sustancias reductoras (DTT, 2-mercaptoetanol)
OPERACIONES
1. Preparación de la curva estándar
- Diluya una solución estándar de BSA de 1 mg/mL con solución salina normal a una serie
concentración. El gradiente de dilución recomendado es el siguiente: 0, 0,2, 0,3, 0,4,
0,6, 0,7, 0,9, 1 mg/ml
2. Medida de las muestras
- Pocillo estándar: Añadir 20 μl de solución estándar a diferentes concentraciones
- Pocillo de muestra: Añadir 20 μl de muestra
- Añadir 200 μl de la solución BCA a cada uno de los pocillos anteriores.
- Mezclar en el vórtex durante 20 segundos
- Incubar a 37ºC durante 30 minutos
- Medir el valor OD para cada pocillo a 562 nm con el lector de microplacas
CÁLCULO DE LA CANTIDAD DE PROTEÍNA
La cantidad de proteína se calculará a partir de la ecuación 3, después de realizar una recta de calibrado, donde se represente el valor OD estándar frente a las concentraciones crecientes conocidas de BSA.
– a: pendiente de la curva estándar
– b: punto de corte con el eje y en la curva estándar
– ΔA562=ODmuestra – ODblanco
– f: factor de dilución de la muestra

OTRAS TÉCNICAS PARA MEDIR LA CANTIDAD DE PROTEÍNA
En la tabla 21 se resumen los principales métodos para la cuantificación de proteínas, así como sus rangos de sensibilidad, sus ventajas e inconvenientes.
| Método | Rango de sensibilidad (μg) | Coeficiente de extinción o cálculo de concentración | Ventajas | Inconvenientes |
| Métodos de absorción | No se pierden las muestras |
Interfieren muchos compuestos que absorben en el UV |
||
| A280 | 100-3000 | ε280 = 1 mL/mg cm | ||
| A205 | 3-100 | ε205 = 31 mL/mg cm | ||
| A280-A260 | 100-3000 | Proteína (mg/mL) = (1.55A280 – 0.76A260) | ||
| A235-A280 | 25-700 | Proteína (mg/mL) = (A235 – A280)/2.51 | ||
| A224-A236 | 5-180 | Proteína (mg/mL) = (A224 – A236)/0.6 | ||
| A215-A225 | 2-45 | Proteína (μg/mL) = 144(A215 – A225) | ||
| Métodos derivados colorimétricos | ||||
| Biuret | 1000-10000 | ε545 = 0.06 mL/mg cm | Bastante específico para proteínas Muestra pocas interferencias Es barato |
Tiene poca sensibilidad |
| Lowry | 25-100 a 500 nm 2-30 a 660 nm 1-2 a 750 nm |
Usar curva estándar | Tiene bastante sensibilidad |
No todas las proteínas reaccionan igual Mustra muchas interferencias como detergentes no iónicos, sulfato amónico etc |
| Bradford | 1-15 | ε595 = 81 mL/mg cm | Muy sensible | Muestra interferencias con detergentes |
| BCA | 0’5-10 | Usar curva estándar | Es el método mas sensible Es el que muestra menos interferencias |
|
| Métodos derivados fluorimétricos | ||||
| o-ftaldehído | 1-5 λexcitación a 340 nm λemisión a 475 nm |
Usar curva estándar | Muy sensible | La interferencia de aminas contaminantes en la muestra No todas las muestras reaccionan igual. |
Esta entrada tiene 0 comentarios