
CUESTIONES PREVIAS
En esta sección se exponen algunos aspectos a tener en cuenta antes de comenzar con los pasos principales del proceso de clonación de la secuencia del gen que codifica la insulina humana.
- ¿Dónde se encuentra el gen de la insulina? El gen que codifica la insulina humana se encuentra en el cromosoma 11 (Ver localización del gen INS)
- ¿Utilizar la secuencia de ADN o ARN? El gen de la insulina se encuentra en el cromosoma 11 de todas las células, pero la expresión de dicho gen se lleva a cabo en órganos internos, como el páncreas. Por lo tanto, el principal problema de utilizar ARN para la clonación de este gen es la necesidad de acceder a las células pancreáticas, donde tiene lugar su expresión, lo cual complicaría la metodología.
Además, el ARNm es muy inestable, y complica el proceso de amplificación. Por otro lado, se puede ser diabético por tener un error en la secuencia promotora del gen, y al utilizar ARNm no podríamos estudiar dicha parte de la secuencia.
Por estas razones usaremos la secuencia de ADN para la clonación del gen de la insulina
- ¿Saliva o sangre? Para la extracción de ADN usaremos sangre
PASO 1: OBTENCIÓN DE LA MUESTRA. EXTRACCIÓN DE ADN DE LA SANGRE
INTRODUCCIÓN
A partir de un kit de purificación de gDNA ChargeSwitch ® vamos a extraer ADN genómico desde pequeños volúmenes de sangre. Los kits de sangre ChargeSwitch ® gDNA están diseñados para permitir el aislamiento de ADN genómico a partir de las siguientes cantidades de sangre humana fresca o congelada tratada con el anticoagulante EDTA o citrato. El ADN genómico purificado es adecuado para su uso en aplicaciones posteriores que incluyen PCR, digestión con enzimas de restricción y transferencia Southern
- ChargeSwitch ® gDNA 20 µl Blood Kits: Purifica hasta 600 ng de ADN genómico de 10-20 µl de sangre humana.
- ChargeSwitch ® gDNA 100 µl Blood Kit: Purifica hasta 3 µg de ADN genómico de 50 a 100 µl de sangre humana.
- ChargeSwitch ® gDNA 1 ml Blood Kit: purifica hasta 20 µg de ADN genómico de 1 ml de sangre humana
Los kits de sangre ChargeSwitch ® gDNA requieren un volumen de muestra y permiten un número de preparaciones diferente (figura 1). Por esta razón, se usará el ChargeSwitch ® gDNA 20 µl Blood Kits, ya que requiere un volumen de sangre fácil de extraer y permite mayor número de preparaciones.

El kit de ChargeSwitch ® elegido (nº catálogo: CS11010) no está disponible en ThermoFisher. Sin embargo, podemos utilizar el kit ChargeSwitch ® con nº de catálogo CS11040 (ChargeSwitch® gDNA 1ml Serum Kit) por 154,77€, ya que contiene los mismos productos que el kit de interés y se basa en el mismo procedimiento.
Nota 1: ChargeSwitch® gDNA 1 ml Serum Kit utiliza muestras de 0.2-1 ml (200µL – 1000µL) por lo que tendremos que diluir 10 veces (nos interesa extraer 10-20µL). ChargeSwitch ® recomienda que se utilice el tampón de elución ChargeSwitch® ( E5 ; Tris-HCl 10 mM, pH 8,5) que se suministra en el kit para eluir el ADN y obtener mejores resultados, debido a que el pH debe mantenerse entre 8,5-9,0.
El precio del kit ChargeSwitch ® con nº de catálogo CS11040 equivale a 50 preparaciones. Sin embargo, el precio de una sola preparación (para una sola clonación) sería de: 3,09€
La tecnología de ChargeSwitch ® se basa en perlas magnéticas que proporcionan una carga de superficie intercambiable que depende del pH del tampón circundante para facilitar la purificación del ácido nucléico.

Como se muestra en la figura 2, a condiciones de pH bajo, las perlas se encuentran cargadas positivamente. De esta manera, las perlas se unen al ácido nucléico (esqueleto del ácido nucléico cargado negativamente), a diferencia de las proteínas y otros contaminantes, por lo que pueden ser retirados en un tampón de lavado acuoso. Para eluir los ácidos nucleicos, la carga en la superficie de la perla se neutraliza elevando el pH a 8,5 usando un tampón de elución bajo en sal. El ADN purificado se eluye instantáneamente en este tampón de elución y está listo para su uso en aplicaciones posteriores.
MATERIALES
- Los componentes proporcionados por ChargeSwitch® gDNA 1 ml Serum Kit se muestran en la figura 3.

Serum Kit [Fuente: ThermoFisher]
- Los componentes no suministrados por el kit son los siguientes:
– Gradilla de separación magnética adecuada para usar con tubos de microcentrífuga de 1,5 ml: Comprar aquí MagnaRack™ (n.º de catálogo CS15000) por 414,00€ (Figura 4).

– Tubos de microcentrifuga estériles de 1,5 ml: Comprar aquí (nº de catálogo 3451PK) por 19,40€
– Mezclador vortex: Comprar aquí (nº de catálogo 88882010) por 388€ (Figura 5)

– Pipetas: Comprar aquí pipeta monocanal de volumen variable (2 a 20 µl) (nº de catálogo 4641180N) por 305,00€
– Lanceta (para extraer la sangre con un pinchazo de forma sencilla): Comprar aquí lanceta de punción (5 a 30 µl) por 22,62€
DESCRIPCIÓN DEL PROCESO DE EXTRACCIÓN DE ADN
A continuación, se describe el protocolo de purificación de ADN genómico a partir de muestras de sangre de 10-20 µl utilizando el kit ChargeSwitch® gDNA 1ml Serum. Para más información visitar la página de ThermoFisher Scientific.
MATERIAL DE PARTIDA
- Volúmen: 10-20 µl de sangre humana
- Tratamiento: tratado con EDTA o citrato
- Estado de la muestra: Fresco o congelado
ANTES DE COMENZAR
Realice lo siguiente antes de comenzar:
- Prepare una Lysis Mix: Para cada muestra, mezcle 0,5 ml de ChargeSwitch ® Lysis Buffer (L12) y 5 µl de Proteinasa K para preparar la Lysis Mix. Si está aislando ADN de varias muestras, puede aumentar el volumen de reactivos utilizados y preparar una mezcla maestra de lisis.
- Vórtice el tubo que contiene las microesferas magnéticas ChargeSwitch ® para resuspender por completo y distribuir uniformemente las microesferas en el tampón de almacenamiento.
- Prepare una mezcla de purificación: Para cada muestra, mezcle 20 µl de microesferas magnéticas ChargeSwitch® (totalmente resuspendidas; consulte más arriba) y 100 µl de tampón de purificación ChargeSwitch® (N5) para preparar la mezcla de purificación. Si está aislando ADN de varias muestras, puede aumentar el volumen de reactivos utilizados y preparar una mezcla maestra de purificación.
PREPARACIÓN DE LISADO
Siga el procedimiento que se indica a continuación para preparar un lisado a partir de 10-20 µl de muestra de sangre.
- Transfiera la muestra de sangre de 10-20 µl a un tubo de microcentrífuga estéril (o una placa de pocillos profundos de 96 x 2 ml).
- Añada 0,5 ml de Lysis Mix (ver arriba) a la muestra y pipetee hacia arriba y hacia abajo suavemente 5 veces para mezclar.
Nota 2: utilice una punta de pipeta de 1 ml ajustada a 450 µl para mezclar la muestra. Asegúrese de que la punta esté sumergida y pipetee hacia arriba y hacia abajo suavemente para evitar la formación de burbujas. - Incube la muestra a temperatura ambiente durante 10 minutos o hasta que la muestra esté transparente y sin grumos visibles.
- Continúe con la unión del ADN.
UNIÓN DEL ADN
Siga el procedimiento que se indica a continuación para unir el ADN a las perlas magnéticas ChargeSwitch ®.
- Pipetee suavemente hacia arriba y hacia abajo la mezcla de purificación que contiene las perlas magnéticas ChargeSwitch ® para volver a suspender completamente las perlas.
- Agregue 120 µl de mezcla de purificación ChargeSwitch ® a la muestra digerida (del paso 3 anterior) y pipetee hacia arriba y hacia abajo suavemente 5 veces para mezclar.
Nota 3: utilice una punta de pipeta de 1 ml ajustada a 550 µl para mezclar la muestra. Asegúrese de que la punta esté sumergida y pipetee hacia arriba y hacia abajo suavemente para evitar la formación de burbujas. - Incube a temperatura ambiente durante 1 minuto para permitir que el ADN se una a las perlas magnéticas ChargeSwitch® .
- Coloque la muestra en el MagnaRack™ (o en el separador magnético de 96 pocillos si utiliza una placa de pocillos profundos de 96 pocillos) durante 1 minuto o hasta que las perlas hayan formado un sedimento compacto.
- Sin retirar la muestra del MagnaRack™, retire con cuidado el sobrenadante y deséchelo. Tenga cuidado de no perturbar el sedimento de perlas inclinando la pipeta de manera que la punta apunte lejos del sedimento (figura 6).
- Retire la muestra que contiene las perlas magnéticas sedimentadas del MagnaRack™. No debe haber sobrenadante en el tubo.
- Añada 500 µl de tampón de lisis ChargeSwitch ® (L12; sin proteinasa K) al tubo y pipetee hacia arriba y hacia abajo suavemente 3 veces para mezclar. Use una punta de pipeta de 1 ml ajustada a 450 l.
- Agregue 50 µl de tampón de purificación ChargeSwitch ® (N5) y pipetee hacia arriba y hacia abajo suavemente 3 veces para mezclar. Use una punta de pipeta de 1 ml ajustada a 500 l.
- Incubar a temperatura ambiente durante 1 minuto.
- Coloque la muestra en el MagnaRack™ (o en el separador magnético de 96 pocillos, si corresponde) durante 1 minuto o hasta que las perlas hayan formado un sedimento compacto.
- Sin retirar la muestra del MagnaRack™, retire con cuidado el sobrenadante y deséchelo. Tenga cuidado de no perturbar el sedimento de perlas inclinando la pipeta de manera que la punta apunte hacia el lado contrario del sedimento.
- Proceda inmediatamente a Lavado de ADN, a continuación.

LAVADO DE ADN
- Retire la muestra que contiene las perlas magnéticas sedimentadas del MagnaRack™ (Paso 11, arriba). No debe haber sobrenadante en el tubo.
- Agregue 500 µl de tampón de lavado ChargeSwitch ® (W12) a la muestra y pipetee hacia arriba y hacia abajo suavemente dos veces para volver a suspender las perlas magnéticas.
Nota 4: Use una punta de pipeta de 1 ml ajustada a 900 µl para mezclar la muestra. Asegúrese de que la punta esté sumergida y pipetee hacia arriba y hacia abajo suavemente para evitar la formación de burbujas. - Coloque la muestra en el MagnaRack™ durante 1 minuto o hasta que las perlas hayan formado un sedimento compacto.
- Sin retirar la muestra del MagnaRack™, retire con cuidado el sobrenadante y deséchelo. Tenga cuidado de no perturbar el sedimento de perlas inclinando la pipeta de manera que la punta apunte hacia el lado contrario del sedimento.
- Continúe con Elución de ADN.
ELUCIÓN DE ADN
- Retire la muestra que contiene las perlas magnéticas sedimentadas del MagnaRack™ (Paso 4, arriba). No debe haber sobrenadante en el tubo.
- Agregue 100 µl de tampón de elución ChargeSwitch ® (E5) (o tampón TE, pH 8,5) a la muestra y pipetee hacia arriba y hacia abajo suavemente 10 veces para volver a suspender las perlas magnéticas.
Nota 5: No utilice agua para la elución. El ADN no eluirá debido a la escasa capacidad amortiguadora del agua. - Incubar a temperatura ambiente durante 1 minuto.
- Coloque la muestra en el MagnaRack™ durante 3 minutos o hasta que las perlas hayan formado un sedimento compacto.
- Sin retirar el tubo del MagnaRack™, extraiga con cuidado el sobrenadante que contiene el ADN y colóquelo en un tubo de microcentrífuga estéril (o en una placa de microtitulación con fondo en U de 96 x 300 µl). Tenga cuidado de no perturbar el sedimento de perlas inclinando la pipeta de manera que la punta apunte hacia el lado contrario del sedimento.
- Deseche las perlas magnéticas usadas. No reutilice las cuentas.
ALMACENAMIENTO DE ADN
Almacene el ADN purificado a -20 °C o utilícelo inmediatamente para el análisis posterior. Anule la congelación y descongelación repetidas veces del ADN.
PASO 2. DISEÑO DE CEBADORES
INTRODUCCIÓN
En esta sección se describen algunas consideraciones para el diseño de cebadores. Se diseñarán cebadores para amplificar el ADN.
- Región codificante del gen
Aunque algunas enfermedades, como la diabetes, puedan ser debido a errores en la región reguladora extragénica o en la secuencia promotora del gen que codifica la insulina, éstas no serán el objeto de estudio: este trabajo se centra en la región codificante del gen INS.
De esta manera, se tendrá en cuenta la región CDS, la cual representa la secuencia del gen que se transcribirá y, por lo tanto, la que interesará amplificar.
Sgún la base de datos NCBI, el gen que codifica para la insulina (GenBank: AH002844.2) presenta el siguiente intervalo de CDS:
– 2424…2610 (187 bases)
– 3397…3542 (146 bases)
Por lo tanto, se diseñarán 4 cebadores teniendo en cuenta el rango de la región codificante del gen
- Afinidad por otros sitios de unión
Hay que tener en cuenta que los cebadores pueden tener afinidad por otros sitios de unión dentro de la secuencia que queremos amplificar. Por lo tanto, si los cebadores diseñados presentan esta característica, vamos a priorizar aquellos que tengan más errores en el extremo 3′ que en el 5′ de la secuencia.
Esto se debe a que la Taq polimerasa se encarga de amplificar la secuencia de nucleótidos desde un extremo 3′. Por lo tanto, cuando encuentre un extremo 3′ erróneo se hará más complicada su unión y, por lo tanto, su amplificación. De esta manera, se evita la amplificación de extremos no deseados.
- Temperatura de fusión (Tm)
La temperatura de fusión (Tm) es la temperatura en la cual el 50% del ADN tiene sus hebras separadas (desnaturalización del ADN).
La temperatura de fusión condiciona el diseño de cebadores, así como la diferencia entre la Tm de los cebadores y la Tm del ADN. Con una diferencia de Tm entre 15-20º conseguiremos que el cebador compita con el producto (ADN) y no se pegue el producto entre sí.
Por lo tanto, antes de comenzar con el diseño de cebadores, tenemos que calcular la Tm del ADN.
CÁLCULO DE LA Tm DE ADN
- Existen algunas páginas web que calculan la Tm del ADN, como ENDMEMO, EMBOSS, etc. Sin embargo, para la mayoría es necesario conocer la concentración del ADN problema.
Nanodrop es un dispositivo capaz de medir pequeños volúmenes de ADN, ARN o proteínas, y sirve para cuantificar de manera exacta y fiable muestras que no son fácilmente medibles con otros utensilios. A continuación se muestra un vídeo que ilustra el procedimiento de Nanodrop.
En ThermoFisher Scientific podemos encontrar diferentes dispositivos para la cuantificación de ARN/ADN a través de espectroscopía UV-visible, los cuales incluyen el espectrofotómetro NanoDrop One para un análisis práctico de microvolúmenes de una sola muestra (nº de catálogo ND-ONE-W) por 10.850€. Este dispositivo permite el uso de concentraciones bajas 2,0 ng/µl de ADNdc.
Algunas bibliotecas estándar de Illumina, como Nextera, requieren el uso de métodos de tinción fluorescente específicos de dsDNA para una cuantificación precisa. Estos métodos suelen medir la concentración de dsDNA en ng/µl. Para convertir de ng/µl a nM puede seguirse la siguiente ecuación:

Asumiendo que Nanodrop mide 2 nanogramos y que el protocolo de PCR qur utilizaremos para amplificar la secuencia se realiza en un volumen de 25 µl (Ver paso 3. AMPLIFICACIÓN DEL ADN). Además, la región codificante del ADN problema contiene 333 pb (CDS 1 = 187 pb; CDS 2 = 146 pb):
(2 ng/ 25 µl) / (660 g/mol x 333 pb) x 106= 0,364 nM es la concentración de ADN
Una vez conocida la concentración de ADN, podemos calcular la Tm del ADN (en concreto calcularé la Tm de cada región codificante: CDS 1 y CDS 2) a través de EMBOSS, la cual necesita los datos de la concentración de iones (50mM de K+) que se utilizará en el proceso de amplificación del ADN por PCR (Ver paso 3. AMPLIFICACIÓN DEL ADN).
– Tm de la ventana de 187 nucleótidos: 93,3º
– Tm de la ventana de 146 nucleótidos: 90,8º
Teniendo en cuenta que EMBOSS aumenta alrededor de 15º la Tm:
– Tm de la ventana de 187 nucleótidos: (93,3 – 15) = 78,3º
– Tm de la ventana de 146 nucleótidos: (90,8 – 15) = 75,8º
Una vez conocida la Tm del ADN, podemos determinar la Tm de los cebadores con la consideración anteriormente mencionada: Tm ADN – Tm cebador más inestable = 15-20º
– Tm cebador para CDS 1: (78,3-15) = 63,3º
– Tm cebador para CDS 2: (75,8-15) = 60,8º
- Otra forma de determinar la Tm del ADN, para obtener la Tm de las regiones codificantes, puede ser mediante la página Oligo.net
En este sitio web se puede descargar la versión 7 de oligos, pero esta es una versión de pago. Una alternativa es acceder al Manual con información sobre el análisis de cebadores y consideraciones para realizar amplificación por PCR.
En dicho manual podemos encontrar la siguiente ecuación (ecuación 2) para obtener la Tm del ADN (en este caso, de las regiones codificantes del gen). Esta alternativa, a diferencia de la anterior, tiene en cuenta el %GC de la secuencia. Por lo tanto, aunque ambas son aproximaciones de Tm, es importante abarcar varias consideraciones.

Al igual que en el caso anterior para el cálculo de Tm, la [K+] es de 50 mM según el protocolo de amplificación de PCR (Ver paso 3. AMPLIFICACIÓN DEL ADN). El %mismatch hace referencia a las bases que no coinciden y que, por lo tanto, no establecen puentes de hidrógeno, por lo que no contribuyen a la Tm. En este caso tenemos un 100% de bases que coinciden, por lo que %mismatch será un término a despreciar. El %GC (calculado mediante Science buddies) y la longitud de la cadena (Len en la fórmula) será algo característico de cada región codificante (CDS 1 y CDS 2) a las cuales calcularemos la Tm:
| [K+] (mM) | Len | %G | %C | Tm (ºC) | |
| CDS 1 | 50 | 187 | 34,75 | 30,48 | 107,93878 |
| CDS 2 | 50 | 146 | 28,08 | 35,6 | 106,55242 |
Una vez conocida la Tm del ADN, podemos determinar la Tm de los cebadores con la consideración anteriormente mencionada: Tm ADN – Tm cebador más inestable = 15-20º
– Tm cebador para CDS 1: (107,93-15) = 92,9º
– Tm cebador para CDS 2: (106,55-15) = 91,55º
- Podemos observar diferentes valores de Tm, dependiendo de la ecuación o base de datos utilizada. Estas diferencias posiblemente sean como consecuencia de las diferentes aproximaciones que se han considerado en cada caso.
En este caso, utilizaremos los valores de Tm de los cebadores que hemos obtenido en la primera aproximación (63,3º y 60,8º) porque es la que más se aproxima a los valores de Tm característicos del genoma humano.
- Es importante mencionar que para estos cálculos se ha utilizado como concentración de sal la que corresponde a la sal KCl del tampón de PCR, en lugar de la concentración de sal MgCl2. Esto se debe principalmente a la despreciable cantidad de sal Mg2+ , a la importancia de la sal K+ (ver enlace), así como a la despreciable cantidad de sal K+ dentro de la Taq DNA polimerasa (ver PASO 3. AMPLIFICACIÓN POR PCR).
PROCEDIMIENTO
Una vez hemos introducido las consideraciones principales para el objetivo de esta sección, damos paso al procedimiento para el diseño y selección de los cebadores que usaremos para amplificar el ADN.
Como ya hemos mencionado, se diseñarán 4 cebadores (dos para cada región codificante CDS 1 y CDS 2), a través de la página NCBI (pick primers).
- Cebadores de CDS 1
– La región codificante CDS 1 tiene una secuencia de 187 nucleótidos en el siguiente intervalo: 2424…2610
– La región codificante CDS 1 tiene una Tm de 78,3º
– Los cebadores de la región codificante CDS 1 tienen una Tm de 63,3º (será la Tm óptima)
 |  |
Con las restricciones de la figura 7 y los datos mencionados, obtenemos los siguientes cebadores (Figura 8):

Como podemos observar con esta vista gráfica, los cebadores reverse (tanto del primer nº1 como del nº2) presentan una distancia de separación con CDS 1 de muy pocos nucleótidos (alrededor de 20 nucleótidos). Por lo tanto, en el momento de secuenciar pueden verse señales contaminadas, ya que las 25-30 primeras bases no se pueden secuenciar bien por el método que llevaré a cabo en este protocolo (Ver paso 4. SECUENCIACIÓN DEL ADN)
Por esta razón, abriré la ventana de restricciones tal y como muestra la figura 9.
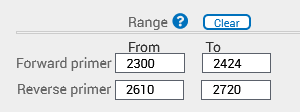 |  |
Con las restricciones de la figura 9 y los datos mencionados, obtenemos los siguientes cebadores (Figura 10)

En la figura 10 podemos ver que al abrir la ventana de nucleótidos (en concreto en el cebador reverse), así como aumentar la diferencia de Tm y disminuir un 1ºC el valor de Tm óptima, aparece mayor número de posibles cebadores (8 más).
Sin embargo, la mayoría presentan alto grado de complementariedad sobre sí mismos en el extremo 3′, muchas regiones complementarias no deseadas, un producto demasiado largo (que será un inconveniente en el momento de secuenciar) y valores de Tm demasiado alejados del valor deseado.
A pesar de estos inconvenientes, el cebador nº7 de la figura 10 (figura 11) presenta mayores ventajas que los cebadores de la figura 8 con las anteriores limitaciones y consideraciones. Este cebador no proporciona un producto demasiado largo, y las cadenas con las que presenta complementariedad tienen más errores en el extremo 3′ que en el 5′. Además, tiene una distancia de separación con la región CDS 1 que permite descartar las regiones mal secuenciadas. Por otro lado, la Tm no se aleja demasiado de la esperada.

- Cebadores de CDS 2
En este apartado seguiremos la misma metodología en el diseño de los cebadores.
– La región codificante CDS 2 tiene una secuencia de 146 nucleótidos en el siguiente intervalo: 3397…3542
– La región codificante CDS 2 tiene una Tm de 75,8º
– Los cebadores de la región codificante CDS 2 tienen una Tm de 60,8º (será la Tm óptima)
 |  |
Con las restricciones de la figura 12 y los datos mencionados, obtenemos los siguientes cebadores (Figura 13)

Como podemos observar a primera vista, todos los cebadores obtenidos con las restricciones descritas en la figura 12 parecen tener una buena distancia de separación con la región CDS 2 y no aportarán señales contaminadas en la secuenciación (Ver PASO 6. SECUENCIACIÓN DEL ADN).
Con una vista más detallada de cada cebador obtenido (no se muestra en la figura) se observa que nignuno presenta complementariedad con otras regiones no deseadas. Sin embargo, algunos pares de cebadores (en concreto algunos cebadores reverse) sí presentan alta complementariedad con la región 3′ de sí mismo.
Como conclusión, considero que el par de cebadores nº1 es el que reúne mayor número de ventajas (figura 14)

PEDIDO DE CEBADORES
Una vez hemos diseñado y seleccionado los 4 cebadores tenemos que realizar el pedido de cebadores. En este caso, se encargarán los cebadores a ThermoFisher Scientific (Tabla 2).
| Forward CDS 1 | Reverse CDS 1 | Forward CDS 2 | Reverse CDS 2 | Total | |
| Precio (€) | 3,57 | 3,06 | 3,74 | 3,57 | 13,94 |
| Cantidad garantizada (nmol) | 9,9 | 4,9 | 9,3 | 9,1 | 33,2 |
| Cantidad pedida (nmol) | 25 | 25 | 25 | 25 | 100 |
PASO 3. AMPLIFICACIÓN DEL ADN. Polymerase Chain Reaction (PCR)
INTRODUCCIÓN
La PCR se ha convertido en una técnica común en muchos laboratorios de biología molecular. Debido a que cada reacción es un experimento único, las condiciones óptimas requeridas para generar un producto varían. Comprender las variables en una reacción mejorará en gran medida la eficiencia de la resolución de problemas, lo que aumentará la posibilidad de obtener el resultado deseado.
Existen numerosos protocolos para la amplificación el ADN por PCR, los cuales se basan en el mismo mecanismo: la reacción en cadena de la polimerasa (vídeo 3).
Además del mecanismo, los protocolos de PCR se asemejan en las consideraciones y precauciones que debemos tener en cuenta para el proceso. Éstas se pueden resumir en el vídeo 4.
A pesar de que el vídeo 4 explica un protocolo para realizar la PCR, en este caso se utilizará Thermo Scientific Taq DNA Polymerase with KCl Buffer, ya que, a diferencia del protocolo del vídeo 4, Thermo Scientific Taq DNA polymerase utiliza KCl para la reacción de PCR. Es importante tener en cuenta la sal de KCl en el tampón de PCR (figura 16) porque actúa neutralizando la carga presente en el esqueleto del ADN. Para más información sobre la importancia del KCl en la reacción de PCR, así como la presencia de MgCl2, puede consultarse el siguiente enlace.
La información sobre pedidos puede hacerse siguiendo este enlace, o bien, buscando a través de los códigos aportados por ThermoFisher (figura 15).

MATERIALES
- Los componentes proporcionados por ThermoScientific Taq DNA Polymerase with KCl Buffer se muestran en la figura 16
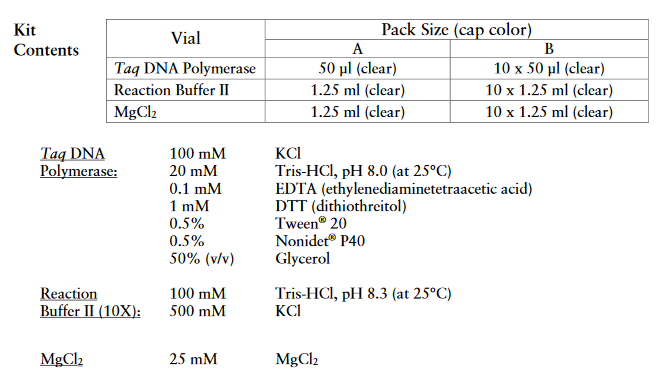
- Los componentes no suministrados por el kit son los siguientes:
– dNTP Mix (20mM): Comprar aquí un set de dNTP (nº de catálogo R0186) por 1.414,00€
– Agua de calidad PCR: Comprar aquí (nº de catálogo W1754-5VL) por 182,00€
– Termociclador: Comprar aquí (nº de catálogo 15806731) por 9698.36€
DESCRIPCIÓN DEL PROCESO DE AMPLIFICACIÓN POR PCR
CÁLCULOS PREVIOS
Antes de comenzar con los pasos descritos en el protocolo para amplificar el ADN, debemos manipular y diluir los cebadores para su uso en el PCR. Tenemos que considerar las concentraciones y volúmenes descritos en el protocolo, mostrados en la figura 17.

Hay que tener en cuenta que, al tener dos exones diferentes, en este trabajo se va a realizar la amplificación de cada CDS por separado, es decir, en diferentes reacciones de PCR. Sin embargo, al tratarse del mismo método para ambos, sólo se describirá el proceso de amplificación por PCR una vez (para el CDS 1), el cual será aplicable para el CDS 2.
- ¿Qué concentración de cebadores hay que preparar?
Para calcular la concentración de solución madre de cebadores que debemos preparar hay que tener en cuenta los siguientes datos:
- Se añaden 1,25 µl (según figura 17)
- La concentración final de cada cebador es de 0,5 µM (según figura 17)
- El tubo de PCR es de 25 µl (según figura 17)
Por lo tanto, la cantidad de cebadores que hay que añadir en 25 µl, para que la concentración de cebadores sea de 0,5 µM es:
1 L = 106 µl –> 0,5 µmol
25 µl –> x µmol
x = 0,0000125 µmol = 12,5 pmol
Estos 12,5 pmol tenemos que añadirlos a 1,25 µl y obtendremos la siguiente concentración de cebadores:
1 L = 106 µl –> x pM (utilizamos 1 L para obtener la concentración directamente)
1,25 µl –>12,5 pmol
x = 10000000 pM = 10 µM
- ¿Cómo preparo 10 µM de cebadores?
La siguiente pregunta hace referencia a la manipulación de los cebadores cuando nos lo entrega la empresa (Ver paso 2. DISEÑO DE CEBADORES). Aquí debemos considerar que, muchas veces, las empresas nos proporcionan una cantidad de cebadores que no se corresponde a la que ellos nos indican. Por eso, sería necesaria una cuantificación previa utilizando Nanodrop (vídeo 2).
Para preparar una concentración de 10 µM de cada cebador (tanto para el CDS 1 como para el CDS 2) hay que tener en cuenta los siguientes datos:
- Tenemos 25 nmol de cebadores (según tabla 2)
- Los tubos albergan como máximo 1,5 ml
- Queremos preparar 10 µM de cada cebador (en total hay que preparar 4 cebadores)
10 µmol = 104 nmol –> 103 ml
25 nmol –> x ml
x = 2,5 ml
Sin embargo, los tubos no pueden albergar 2,5 ml por lo que tendríamos que hacer la siguiente consideración: En lugar de echar 2,5 ml (2500 µl) de agua al tubo, añadiremos 0,25 ml (250 µl), de manera que obtendremos un tubo 10 veces más concentrado. Podemos comprobarlo de la siguiente manera:
250 µl –> 25 nmol
106 µl –> 100000 nM = 100 µM
Para solucionar el aumento de concentración en el tubo, podemos diluir 10 veces añadiendo 10μL de
solución madre a 90μL de tampón (obtendríamos así 10μM). Esto lo podemos distribuir en un total de 10 tubos (dilución 10+90). Así, si se contamina algún tubo o cometemos algún error, tenemos margen para deshechar algún tubo.
- ¿Cuántas reacciones de PCR podemos llegar a realizar?
- Tubo de PCR de 25 µl (según figura 17)
- La concentración final de cada cebador es de 0,5 µM (según figura 17)
- Hay que añadir 12,5 pmol de cada cebador en los tubos (según cálculos anteriores)
25 nmol = 25000 pmol en total
25000 pmol en total / 12,5 pmol en cada tubo = 2000 reacciones posibles
PASOS DEL PROTOCOLO DE PCR
Una vez realizados los cálculos, podemos seguir los pasos que se describen en el protocolo de PCR (figura 18). Se realizará dos veces el protocolo: una para el CDS 1 y otra para el CDS 2.

RECOMENDACIONES
– Usar un gradiente de temperatura de los tubos en la primera reacción de PCR , para poder seleccionar aquella temperatura con la que se hayan obtenido mejores resultados
– Utilizar puntas de pipetas esterilizadas con radiación gamma, así como pinzas, guantes y gorros esterilizados, para no contaminar las muestras. En este enlace se pueden conseguir puntas desechables esterilizadas con filtro.
– Mantener en hielo los reactivos de la PCR, así como los pocillos, como se muestra en la figura 19.

– Para más información sobre cómo hacer un protocolo básico de PCR, así como algunas consideraciones y errores comunes, pinchar este enlace.
- Importancia del buffer
Un componente esencial en el proceso de extracción de ADN, elución, almacenamiento o electroforesis en gel es un buffer. Los más utilizados suelen ser TBE (Tris-borato-EDTA), TAE (Tris-acetato-EDTA) o TE (Tris-EDTA).
Generalmente, se requiere estabilizar la concentración de H + in vitro sin afectar el funcionamiento del sistema. Un tampón o buffer mantiene constante el pH de una solución absorbiendo los protones que se liberan durante las reacciones o liberando protones cuando son consumidos por las reacciones. Sin embargo, estos tampones mencionados anteriormente presentan diferentes características que debemos considerar, de acuerdo a su composición química:
– TBE: Tris-borato-EDTA
TBE es un buen medio conductor si se compara con TAE, por lo que minimiza el sobrecalentamiento. El borato resuelve mejor los fragmentos <2kb, así que suele usarse TBE para fragmentos más pequeños. El borato es un inhibidor de enzimas, por lo que TBE no es un buen tampón para aislar el ADN con posteriores pasos enzimáticos. Por ejemplo, el arrastre de borato podría afectar a los ligandos de la enzimar.
– TAE: Tris-acetate-EDTA
El acetato mejora la separación de fragmentos grandes de ADN. El TAE es el buffer comúnmente empleado para la separación de fragmentos de DNA por electroforésis. Sus aplicaciones electroforéticas incluyen al análisis de productos de PCR, protocolos de purificación de DNA y experimentos de clonación de DNA. Este buffer posee una fuerza iónica baja al igual que una pobre capacidad de amortiguación de pH. Una de las desventajas del TAE como buffer de electroforesis es que no permite reciclarlo por más de tres corridas electroforéticas seguidas, a diferencia del TBE cuya efectividad permanece inalterada durante 3 o 4 días, sin importar el número de corridas electroforéticas realizadas.
– TE: Tris-EDTA
El tampón TE es utilizado por su eficacia para disolver el ADN y evitar su degradación. Además, tiene un papel importante en el lavado y aislamiento del ADN. Se utiliza en la extracción de ADN porque ayuda en la lisis de la pared celular y la membrana nuclear, manteniendo el pH óptimo de la disolución.
Una de las principales ventajas es que no presenta acetato, que es un inhibidor enzimático. Por lo general, usamos enzimas en el proceso de extracción de ADN y PCR, por lo que si el acetato está presente, inhibirá estos procesos.
Utilizaremos TE preferentemente en los procesos de extracción de ADN, dilución de cebadores, reacción de PCR, electroforesis en gel, etc.
PASO 5. PURIFICACIÓN DEL ADN. ELECTROFORESIS, GEL DE AGAROSA Y AISLAMIENTO
INTRODUCCIÓN
La electroforesis en gel de agarosa es la forma más efectiva de separar fragmentos de ADN de diferentes tamaños que van desde 100 pb hasta 25 kb.
Como se describe en (Checas-Rojas, A., 2017), la electroforesis es un método de separación de biomoléculas como el ADN o ARN de acuerdo a su tamaño, permitiendo además su aislamiento cortando la zona de interés. La metodología básica de electroforesis en ácidos nucléicos consiste en la tinción de ADN (o ARN, si fuera el caso) mediante un compuesto fluorescente (bromuro de etidio), que hace visible a la biomolécula deseada (ADN o, en su caso, ARN) mediante emisión de luz ultravioleta (luz UV). Al aplicar una corriente eléctrica sobre el gel de agarosa (donde previamente hemos introducido nuestra biomolécula), el ADN migrará en sentido opuesto a su carga, es decir, hacia el polo positivo (ánodo).
- El fundamento teórico de la electroforesis y sus aplicaciones pueden visualizarse en el vídeo 5.
- ¿Cómo hacer una electroforesis?
Aunque ya se ha introducido la metodología necesaria para hacer una electroforesis, resulta más fácil visualizarla paso a paso (vídeo 6)
- ¿Cómo hacer un gel de agarosa?
La agarosa es un polímero lineal de galactosa y 3,6-anhidrogalactosa soluble a altas temperaturas (65ºC). Cuando la agarosa se enfría y se forma el gel, los polímeros de agarosa se asocian de forma no covalente y forman una red de haces cuyos tamaños de poro determinan las propiedades de tamizado molecular de un gel. El uso de la electroforesis en gel de agarosa revolucionó la separación del ADN. Antes de la adopción de los geles de agarosa, el ADN se separaba principalmente mediante centrifugación en gradiente de densidad de sacarosa, que solo proporcionaba una aproximación del tamaño
En el vídeo 7 se representa paso a paso cómo prepara un gel de agarosa en el laboratorio. Tal y como explica el vídeo, para disolver la agarosa en el buffer necesitamos calentar la disolución, por lo que debemos extremar las precauciones.
MATERIALES
En este enlace podemos encontrar las tablas de materiales necesarios para realizar una electroforesis en gel de agarosa, así como las cantidades recomendadas según el tamaño de ADN.
Antes de describir los materiales y cantidades necesarias, debemos realizar la elección del porcentaje de gel de agarosa, según el tamaño del fragmento de ADN, ya que esto determinará al resto de componentes (Tabla 3).

Se utilizará un pocentaje de 1,5% para cada electroforesis (para CDS 1 y CDS 2)
Los materiales y componentes necesarios que se describirán serán los necesarios para realizar una electroforesis sencilla y minimizando los componentes carcinógenos. Es decir, aquellos que se requieren para el segundo protocolo de electroforesis descrito (Danagen):
– Agarosa para 100 preparaciones: Conseguir aquí (consultar el precio con Danagen)
– Cubetas y fuentes de electroforesis: Conseguir aquí (consultar el precio con Danagen)
– Marcadores de peso molecular: Conseguir aquí (consultar el precio con Danagen). Se comprarán teniendo en cuenta el tamaño de nuestra muestra
– Tinción de ADN (GELSAFE): Conseguir aquí (consultar el precio con Danagen). GELSAFE Nucleic Acid Gel Stain Solution (20,000x) es una nueva y segura tinción de ácidos nucleicos, alternativa NO TÓXICA al tradicional bromuro de etidio para la tinción de ácidos nucleicos.
– Tampones: Conseguir aquí (consultar el precio con Danagen)
Como alternativa a estos componentes, Danagen proporciona un kit de reactivos de electroforesis (no incluyen las cubetas, los macradores ni la fuente de electroforesis). Este kit (ELECKIT) se puede conseguir aquí, y contiene los siguientes componentes:

PROCEDIMIENTO
A continuación se describen dos protocolos para la separación de fragmentos de ADN mediante electroforesis en gel de agarosa. El motivo de describir dos protocolos es que el primero es un protocolo más complejo y completo. El segundo protocolo utiliza reactivos y componentes diferentes que facilitan algunos pasos del mismo procedimiento (como la preparación del gel o su manipulación, ya que utiliza componentes que no son carcinógenos).
Se realizará un proceso de electroforesis en el que obtendremos 10 pocillos, 5 para los fragmentos del CDS 1 y 5 para los fragmentos del CDS 2. Entre los primeros 5 pocillos y los últimos 5 pocillos, introduciremos los marcadores de peso molecular.
- En esta sección se puede seguir el protocolo descrito por NCBI para la separación de fragmentos de ADN mediante electroforesis en gel de agarosa.
PREPARACIÓN DEL GEL
- Pesar la masa adecuada de agarosa en un matraz Erlenmeyer. Los geles de agarosa se preparan utilizando una solución porcentual p/v. La concentración de agarosa en un gel dependerá del tamaño de los fragmentos de ADN que se van a separar, y la mayoría de los geles oscilan entre el 0,5 % y el 2 %. El volumen del tampón no debe ser superior a 1/3 de la capacidad del matraz.
- Agregue tampón de ejecución al matraz que contiene agarosa. Agitar para mezclar. Los tampones de ejecución de gel más comunes son TAE (Tris-acetato 40 mM, EDTA 1 mM) y TBE (Tris-borato 45 mM, EDTA 1 mM).
- Derretir la mezcla de agarosa/tampón. Esto se hace más comúnmente calentando en un microondas, pero también se puede hacer sobre una llama Bunsen. A intervalos de 30 s, retire el matraz y agite el contenido para mezclarlo bien. Repita hasta que la agarosa se haya disuelto por completo.
- Añadir bromuro de etidio (EtBr) a una concentración de 0,5 μg/ml. Como alternativa, el gel también se puede teñir después de la electroforesis en un tampón de ejecución que contiene 0,5 μg/ml de EtBr durante 15 a 30 minutos, seguido de una decoloración en el tampón de ejecución durante el mismo período de tiempo.
- Deje que la agarosa se enfríe en la mesa de trabajo o mediante incubación en un baño de agua a 65 °C. Si no lo hace, la bandeja de gel se deformará.
- Coloque la bandeja de gel en el aparato de fundición. Alternativamente, también se pueden pegar los bordes abiertos de una bandeja de gel para crear un molde. Coloque un peine apropiado en el molde de gel para crear los pocillos.
- Vierta la agarosa fundida en el molde de gel. Permita que la agarosa se asiente a temperatura ambiente. Retire el peine y coloque el gel en la caja de gel. Alternativamente, el gel también se puede envolver en una envoltura de plástico y almacenar a 4 °C hasta su uso (Figura 20).
Nota 6: EtBr es un cancerígeno sospechoso y debe eliminarse adecuadamente según las normas de la institución. Siempre se deben usar guantes cuando se manipulan geles que contienen EtBr. Están disponibles tintes alternativos para la tinción de ADN; sin embargo, EtBr sigue siendo el más popular debido a su sensibilidad y costo.

CONFIGURACIÓN DE APARATO DE GEL Y SEPARACIÓN DE FRAGMENTOS DE ADN
- Añadir colorante de carga a las muestras de ADN que se van a separar (Figura 21). El colorante de carga de gel se prepara normalmente a una concentración de 6X (0,25 % de azul de bromofenol, 0,25 % de xileno cianol, 30 % de glicerol). La carga de tinte ayuda a rastrear qué tan lejos ha viajado su muestra de ADN y también permite que la muestra se hunda en el gel.
- Programe la fuente de alimentación al voltaje deseado (1-5V/cm entre electrodos).
- Agregue suficiente tampón de corrida para cubrir la superficie del gel. Es importante utilizar el mismo tampón de ejecución que el utilizado para preparar el gel.
- Conecte los cables de la caja de gel a la fuente de alimentación. Encienda la fuente de alimentación y verifique que tanto la caja de gel como la fuente de alimentación estén funcionando.
- Retire la tapa. Lenta y cuidadosamente, cargue la(s) muestra(s) de ADN en el gel (Figura 22). Siempre se debe cargar un marcador de tamaño de ADN adecuado junto con las muestras experimentales.
- Vuelva a colocar la tapa de la caja de gel. El cátodo (conductores negros) debe estar más cerca de los pozos que el ánodo (conductores rojos). Vuelva a verificar que los electrodos estén enchufados en las ranuras correctas de la fuente de alimentación.
- Conectar la alimentación. Corre el gel hasta que el tinte haya migrado a una distancia adecuada.


OBSERVACIÓN DE FRAGMENTOS DE ADN SEPARADOS
- Cuando se haya completado la electroforesis, apague la fuente de alimentación y retire la tapa de la caja de gel.
- Retire el gel de la caja de gel. Drene el exceso de tampón de la superficie del gel. Coloque la bandeja de gel sobre toallas de papel para absorber cualquier tampón de funcionamiento adicional.
- Retire el gel de la bandeja de gel y exponga el gel a la luz ultravioleta. Esto se hace más comúnmente usando un sistema de documentación en gel (Figura 23). Las bandas de ADN deben aparecer como bandas fluorescentes de color naranja. Tome una foto del gel.
- Deseche correctamente el gel y el tampón de ejecución según las normas de la institución.

RESULTADOS REPRESENTATIVOS
La figura 24 representa un resultado típico después de la electroforesis en gel de agarosa de productos de PCR. Después de la separación, los fragmentos de ADN resultantes son visibles como bandas claramente definidas. El patrón o escalera de ADN debe separarse hasta un grado que permita la determinación útil de los tamaños de las bandas de muestra. En el ejemplo que se muestra, los fragmentos de ADN de 765 pb, 880 pb y 1022 pb se separan en un gel de agarosa al 1,5 % junto con una escalera de ADN de 2 log

- En esta sección se puede seguir el protocolo descrito por Danagen. El fundamento es el mismo que en el anterior, pero con algunos cambios de componentes que facilitan algunos pasos del procedimiento.
- Añadir la pastilla de DANAGAROSE TABLET (según tabla 5) al volumen necesario de TAE 1X y esperar al menos 2 minutos antes de calentar.

2. Calentar en el microondas a elevada potencia , fundir la agarosa permitiendo una ebullición de 30 segundos aproximadamente.
3. Mezclar bien.
4. Volver a calentar en el microondas hasta que la solución esté transparente.
5. Enfriar la solución a unos 55ºC. A esta temperatura añadir el GELSAFE Nucleic Acid Stain*.
6. Añadir unos 2.5 microlitros de GELSAFE Nucleic Acid Stain por cada 50 ml de gel de agarosa.
7. Permitir que el gel de agarosa se enfríe hasta que se solidifique.
8. Cargar las muestras utilizando el tampón de carga suministrado 6X DNA Loading buffer y realizar la electroforesis.
9. Detectar las bandas bajo iluminación UV
Según el protocolo, es posible que precipite los componentes de este producto debido a su elevada
concentración. Resuspender con vortex o mediante micopipeta cuando haya pasado mucho tiempo desde su último o uso. Además, los filtros rojo/naranjas utilizados para el bromuro de etidio no deben usarse con el GELSAFE, deben utilizarse los mismos filtros que se utilizan con el SYBR Green. También pueden utilizarse filtros amarillos, verdes o filtros de celofán.
También se recomienda ejecutar los geles a un voltaje bajo y constante (~ 10 V/cm) para minimizar los efectos de calentamiento actuales y asimétricos, que pueden inducir artefactos de banda y resolución deficiente.
Hay que tener en cuenta que no veremos las bandas de cebadores: Son fragmentos muy pequeños y no emiten fluorescencia. Es tan pequeña que se han quedado en el tampón.
PASO 6. SECUENCIACIÓN
INTRODUCCIÓN
La secuenciación de ADN es el proceso que determina la secuencia de bases de los nucleótidos (As, Ts, Cs y Gs) de un fragmento de ADN. Dado que la secuencia de ADN confiere la información que utiliza la célula para la fabricación de las moléculas de ARN y proteínas, disponer de la secuencia de ADN es clave para entender cómo funcionan los genomas. La tecnología de secuenciación de ADN se hizo más rápida y menos costosa, a partir del Proyecto del Genoma Humano (secuenciación Sanger).
La secuenciación Sanger utilizada en el Proyecto del Genoma Humano es un método de secuenciación que fue desarrollado por el bioquímico británico Fred Sanger y sus compañeros en 1977. En esta técnica de secuenciación el ADN blanco es copiado muchas veces y se hacen fragmentos de diferentes longitudes. Los nucleótidos fluorescentes que actúan como «terminadores de cadena» marcan los extremos de los fragmentos y permiten la determinación de la secuencia.
Es importante tener en cuenta que la secuenciación de las primeras bases presentará una señal con más calidad que las últimas. Por eso, es necesario tener fragmentos de no más de 800-1000 pb. Por otro lado, las primeras 25-30 pb no se pueden secuenciar nunca, porque están contaminadas con las señales de los precursores de la muestra.
En el vídeo 8 se explica en qué consiste la secuenciación de Sanger de una forma detallada.
PROCEDIMIENTO
Después de purificar el ADN (PASO 5. PURIFICACIÓN DEL ADN) tendremos 3 tubos: 1. Con el ADN purificado desde el gel que contiene las bandas amplificadas; 2. Un tubo con el cebador forward; 3. Un tubo con el cebador reverse. Como la compañía requiere cantidades de ADN muy diluidas y con una pequeña cantidad, las mediciones se pueden hacer con Nanodrop.
El profesor Gabriel Dorado Pérez, de la Universidad de Córdoba, describe las diferentes metodologías para realizar la secuenciación y, en especial, la secuenciación Sanger en el siguiente capítulo.
La Universidad de Córdoba y, en concreto, la Unidad de Genómica (GEN) del Servicio Central de Apoyo a la Investigación (SCAI), proporciona tarifas para la secuenciación de Sanger (Tabla 6)

REQUERIMIENTOS
A continuación, se describe la información proporcionada por el SCAI de la Universidad de Córdoba sobre las muestras y su análisis.
Las muestras para secuenciación Sangerse procesan dos veces en semana, los lunes y los miércoles (salvo incidencias).
Es muy importante que el usuario prepare siempre las muestras diluidas en agua. Las cantidades requeridas de ADN para la reacción son las descritas en la tabla 7. Se debe completar hasta un volumen total de 6 µl en tubos de PCR de 0,2 ml con agua miliQ

- Preparación de muestras
Teniendo en cuenta que teníamos 5 tubos en stock:
– 25 nmol (=25000 pmol) de forward 1 en 250 µl
– 25 nmol (=25000 pmol) de forward 2 en 250 µl
– 25 nmol (=25000 pmol) de reverse 1 en 250 µl
– 25 nmol (=25000 pmol) de reverse 2 en 250 µl
– ADN extraído en el PASO 1. EXTRACCIÓN DEL ADN
Realizaremos los siguientes cálculos para determinar el volumen de cebador que se requiere:
25000 pmol –> 250 µl
3,2 pmol –> x µl
x = 0,032 µl = 32 nl
Comprar aquí pipeta de 0,1 – 2,5 µl
A continuación se representa la cantidad de ADN que debemos mandar a secuenciar:
– Según la tabla 7 se requieren 20 ng/100 pb
– CDS 1 tiene 187 pb
20 ng –> 100 pb
x ng –> 187 pb
x = 37,4 ng
Según figura 17:
125 ng –> 10 µl
37,4 ng –> x µl
x = 2,99 µl
– CDS 2 tiene 146 pb:
20 ng –> 100 pb
x ng –> 146 pb
x = 29,2 ng
Según figura 17:
125 ng –> 10 µl
29,2 ng –> x µl
x = 2,33 µl
Para analizar la secuenciación se puede usar enlaces como Chromas, BioEdit (Libre para PCs), Sequence Scanner Software v1.0, FinchTV (Libre para Linux, PCs, y MACs), y muchos más.
La empresa nos dará una secuencia en cromatograma (figura 25 y 26) y una secuencia FASTA.


FIN. APROXIMACIÓN DE PRESUPUESTO DEL PROYECTO DE CLONACIÓN GLOBAL y valoración personal del trabajo
En este apartado se hace una recopilación de lo que podría llegar a costar este proceso de clonación siguiendo los pasos descritos y, teniendo en cuenta, que se comprarían todos los intrumentos y productos desde cero. Además, para conocer el precio de algunos materiales se necesitaría contactar con el fabricante, por lo que no aparecen a continuación
En las tablas 8, 9, 10 y 11 se muestran los precios disponibles para los diferentes pasos del proceso de clonación del gen de la insulina humana. El precio total del proceso de clonación es de 23.469,51€
| OBTENCIÓN DE MUESTRA | PRECIO (€) |
| Kit Chargeswitch | 157,44 |
| Gradilla de separación | 414 |
| Tubos microcentrifuga | 19,4 |
| Mezclador vortex | 388 |
| Pipetas | 305 |
| Lancetas | 22,62 |
| Total | 1306,46 |
| DISEÑO DE CEBADORES | PRECIO(€) |
| Nanodrop | 10.850 |
| Cebadores | 13,94 |
| Total | 10.863,94 |
| AMPLIFICACIÓN POR PCR | PRECIO(€) |
| dNTP mix | 1414 |
| Agua de PCR | 182 |
| Termociclador | 9698,36 |
| Total | 11.294,36 |
| SECUENCIACIÓN | PRECIO (€) |
| Secuenciación Sanger | 4,75 |
| Total | 4,75 |
Como impresión personal del trabajo me gustaría destacar la importancia que tiene este tipo de proyectos para una persona que se está formando como bioquímico/a. Es importante conocer la teoría que subyace al proceso de replicación, transcripción y traducción de un gen para el manejo de éstos, pero también es necesario aprender a aprender y a buscar soluciones a los problemas que se puedan plantear en un laboratorio. Es imprescindible saber dónde buscar la información, conocer espacios que te proporcionen los utensilios requeridos en los proyectos de investigación, poder plantear protocolos alternativos y saber valorar entre las opciones válidas que se plantean en cualquier desarrollo. En definitiva, es fundamental aprender a ser autosuficiente y, considero que este tipo de trabajos te abre las puertas a entender cómo hacer las cosas desde cero, aunque sea desde el punto de vista teórico.
Por último, me gustaría poder llevar a cabo esta metodología que he planteado para la clonación del gen de la insulina en un laboratorio, donde complementaría el aprendizaje de este trabajo.
Créditos imagen destacada post: Human Insulin PubChem CID: 118984375
Esta entrada tiene 0 comentarios